

Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort
- PMID: 24416410
- PMCID: PMC3885720
- DOI: 10.1371/journal.pone.0085436
Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort
Abstract
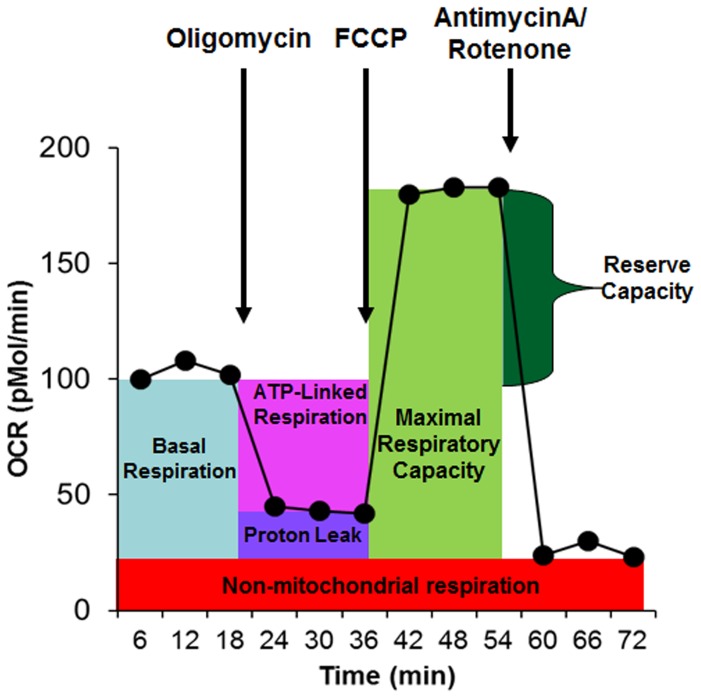
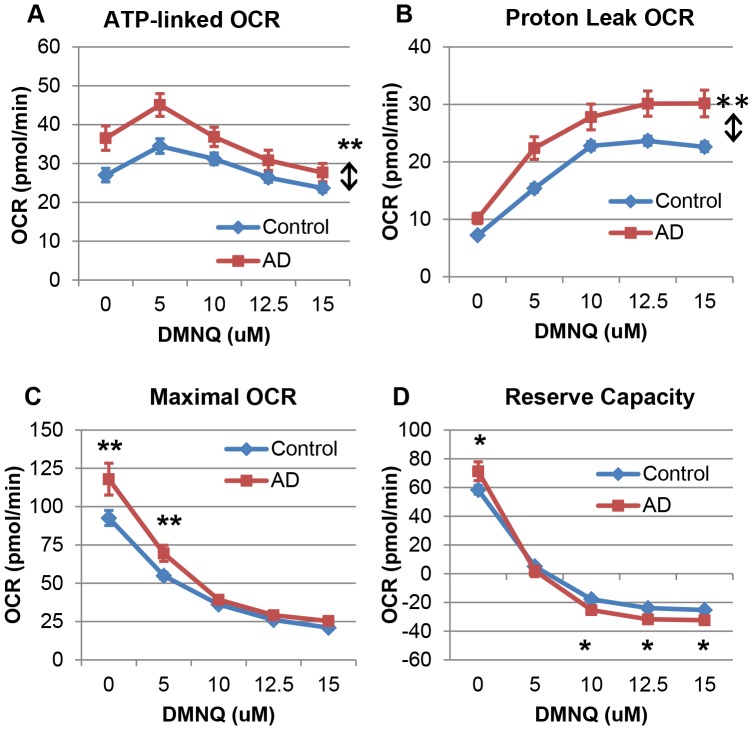
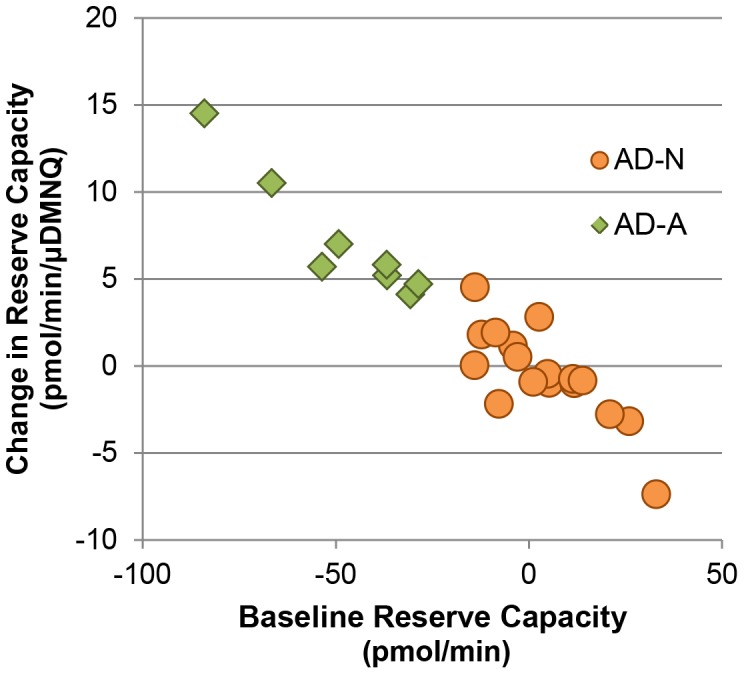
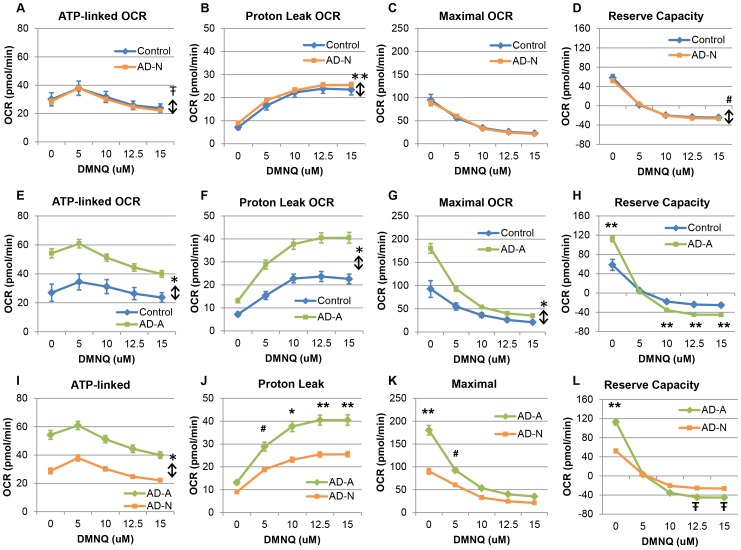
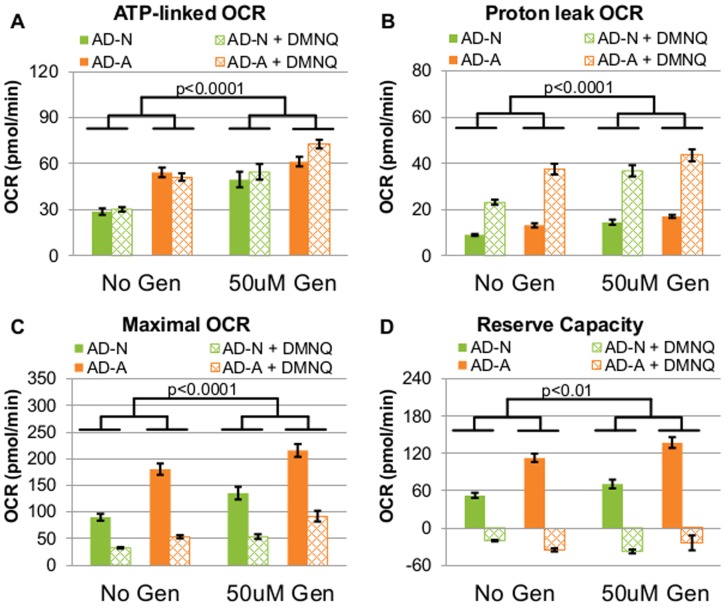
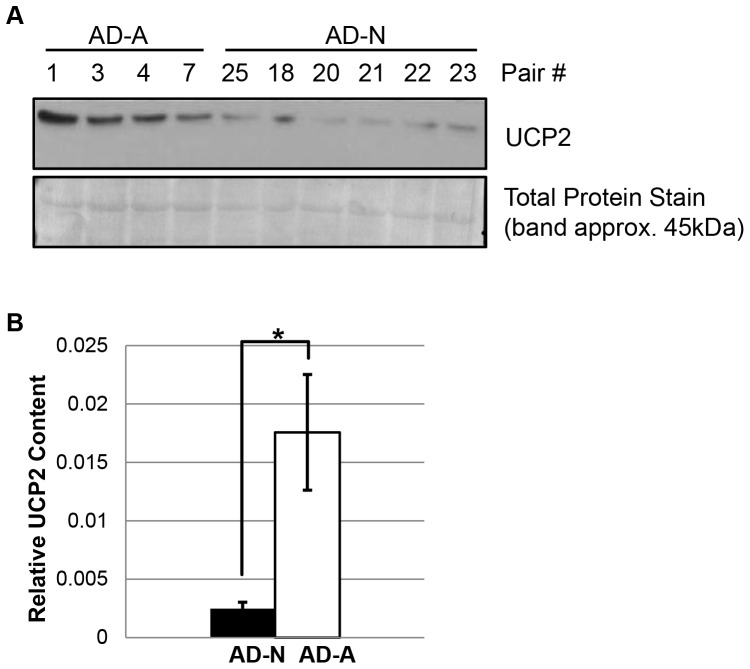
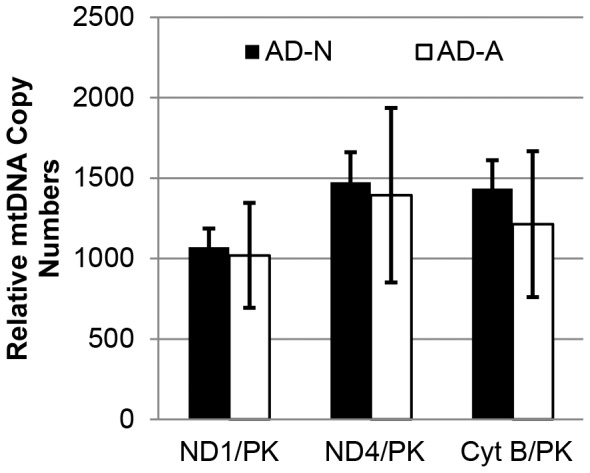
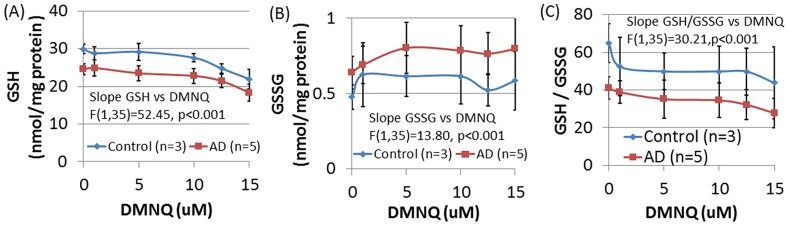
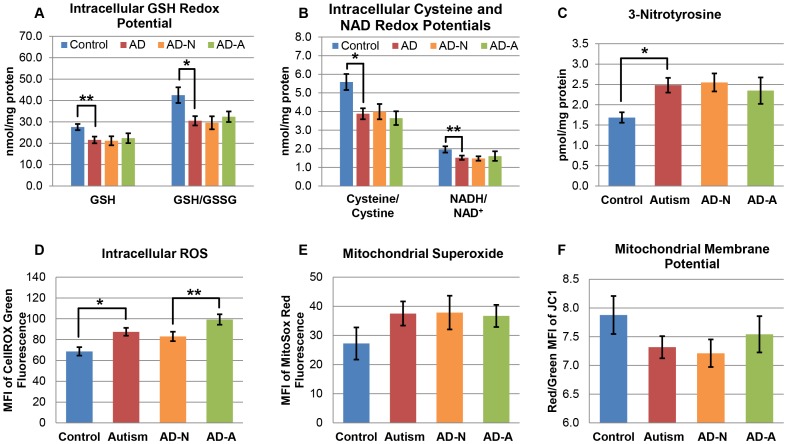
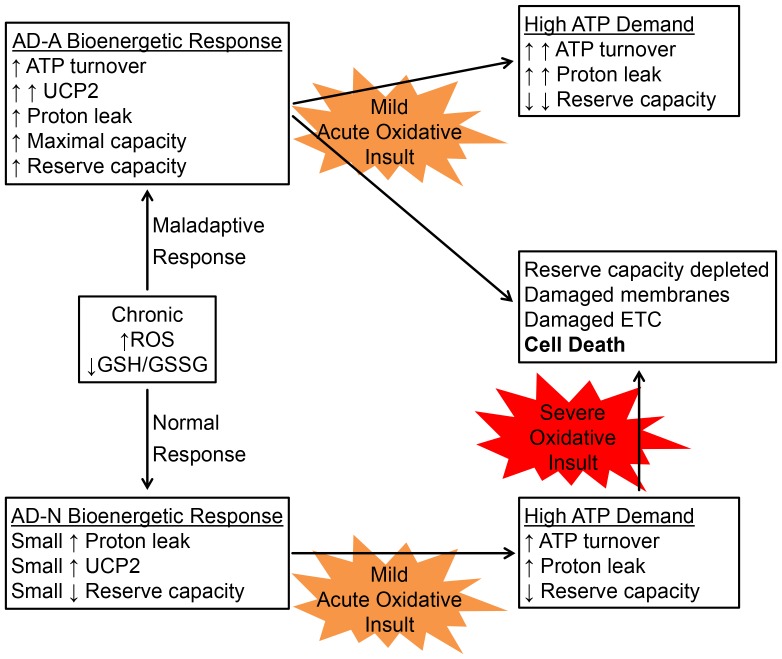
There is increasing recognition that mitochondrial dysfunction is associated with the autism spectrum disorders. However, little attention has been given to the etiology of mitochondrial dysfunction or how mitochondrial abnormalities might interact with other physiological disturbances associated with autism, such as oxidative stress. In the current study we used respirometry to examine reserve capacity, a measure of the mitochondrial ability to respond to physiological stress, in lymphoblastoid cell lines (LCLs) derived from children with autistic disorder (AD) as well as age and gender-matched control LCLs. We demonstrate, for the first time, that LCLs derived from children with AD have an abnormal mitochondrial reserve capacity before and after exposure to increasingly higher concentrations of 2,3-dimethoxy-1,4-napthoquinone (DMNQ), an agent that increases intracellular reactive oxygen species (ROS). Specifically, the AD LCLs exhibit a higher reserve capacity at baseline and a sharper depletion of reserve capacity when ROS exposure is increased, as compared to control LCLs. Detailed investigation indicated that reserve capacity abnormalities seen in AD LCLs were the result of higher ATP-linked respiration and maximal respiratory capacity at baseline combined with a marked increase in proton leak respiration as ROS was increased. We further demonstrate that these reserve capacity abnormalities are driven by a subgroup of eight (32%) of 25 AD LCLs. Additional investigation of this subgroup of AD LCLs with reserve capacity abnormalities revealed that it demonstrated a greater reliance on glycolysis and on uncoupling protein 2 to regulate oxidative stress at the inner mitochondria membrane. This study suggests that a significant subgroup of AD children may have alterations in mitochondrial function which could render them more vulnerable to a pro-oxidant microenvironment derived from intrinsic and extrinsic sources of ROS such as immune activation and pro-oxidant environmental toxicants. These findings are consistent with the notion that AD is caused by a combination of genetic and environmental factors.
Conflict of interest statement
Figures
 p<0.05; ↕ indicates an overall statistical difference between LCL groups.
p<0.05; ↕ indicates an overall statistical difference between LCL groups.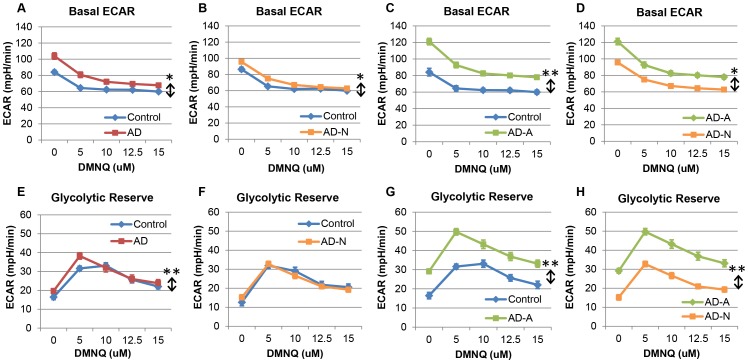
Similar articles
-
Oxidative stress induces mitochondrial dysfunction in a subset of autistic lymphoblastoid cell lines.Transl Psychiatry. 2014 Apr 1;4(4):e377. doi: 10.1038/tp.2014.15. Transl Psychiatry. 2014. PMID: 24690598 Free PMC article.
-
Mitochondrial and redox abnormalities in autism lymphoblastoid cells: a sibling control study.FASEB J. 2017 Mar;31(3):904-909. doi: 10.1096/fj.201601004R. Epub 2016 Nov 18. FASEB J. 2017. PMID: 27864377 Free PMC article.
-
Oxidative Stress Challenge Uncovers Trichloroacetaldehyde Hydrate-Induced Mitoplasticity in Autistic and Control Lymphoblastoid Cell Lines.Sci Rep. 2017 Jun 30;7(1):4478. doi: 10.1038/s41598-017-04821-3. Sci Rep. 2017. PMID: 28667285 Free PMC article.
-
Uncoupling proteins and the control of mitochondrial reactive oxygen species production.Free Radic Biol Med. 2011 Sep 15;51(6):1106-15. doi: 10.1016/j.freeradbiomed.2011.06.022. Epub 2011 Jun 24. Free Radic Biol Med. 2011. PMID: 21762777 Review.
-
Mitochondrial H(+) leak and ROS generation: an odd couple.Free Radic Biol Med. 2005 Jan 1;38(1):12-23. doi: 10.1016/j.freeradbiomed.2004.10.016. Free Radic Biol Med. 2005. PMID: 15589367 Review.
Cited by
-
Massively parallel sequencing of mitochondrial genome in primary open angle glaucoma identifies somatically acquired mitochondrial mutations in ocular tissue.Sci Rep. 2024 Nov 1;14(1):26324. doi: 10.1038/s41598-024-72684-6. Sci Rep. 2024. PMID: 39487142 Free PMC article.
-
Establishment of a platform for measuring mitochondrial oxygen consumption rate for cardiac mitochondrial toxicity.Toxicol Res. 2022 May 10;38(4):511-522. doi: 10.1007/s43188-022-00136-2. eCollection 2022 Oct. Toxicol Res. 2022. PMID: 36277363 Free PMC article.
-
Mitochondria May Mediate Prenatal Environmental Influences in Autism Spectrum Disorder.J Pers Med. 2021 Mar 18;11(3):218. doi: 10.3390/jpm11030218. J Pers Med. 2021. PMID: 33803789 Free PMC article. Review.
-
Modulation of the genome and epigenome of individuals susceptible to autism by environmental risk factors.Int J Mol Sci. 2015 Apr 20;16(4):8699-718. doi: 10.3390/ijms16048699. Int J Mol Sci. 2015. PMID: 25903146 Free PMC article. Review.
-
Mitochondria-containing extracellular vesicles (EV) reduce mouse brain infarct sizes and EV/HSP27 protect ischemic brain endothelial cultures.J Control Release. 2023 Feb;354:368-393. doi: 10.1016/j.jconrel.2023.01.025. Epub 2023 Jan 18. J Control Release. 2023. PMID: 36642252 Free PMC article.
References
-
- APA (1994) Diagnostic and statistical manual of mental disorders. Washington, DC: American Psychiatric Association.
-
- Autism, Developmental Disabilities Monitoring Network Surveillance Year Principal I (2009) Centers for Disease C, Prevention (2009) Prevalence of autism spectrum disorders - Autism and Developmental Disabilities Monitoring Network, United States, 2006. MMWR Surveill Summ 58: 1–20. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
