

The capable ABL: what is its biological function?
- PMID: 24421390
- PMCID: PMC3993570
- DOI: 10.1128/MCB.01454-13
The capable ABL: what is its biological function?
Abstract
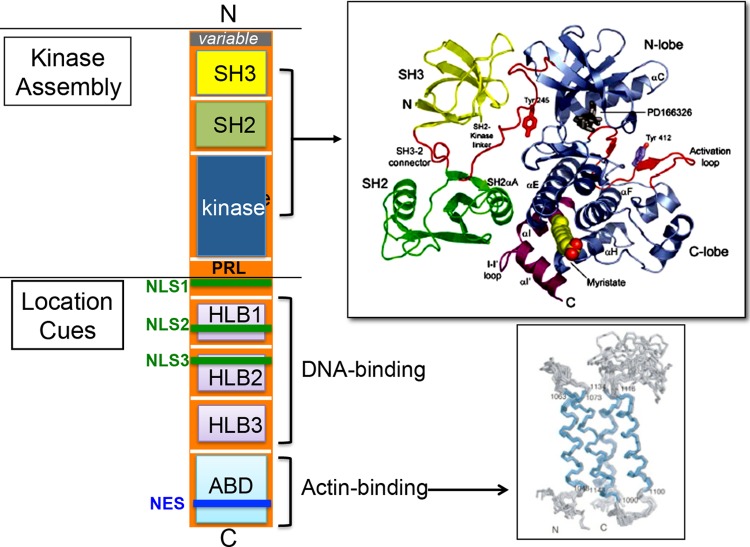
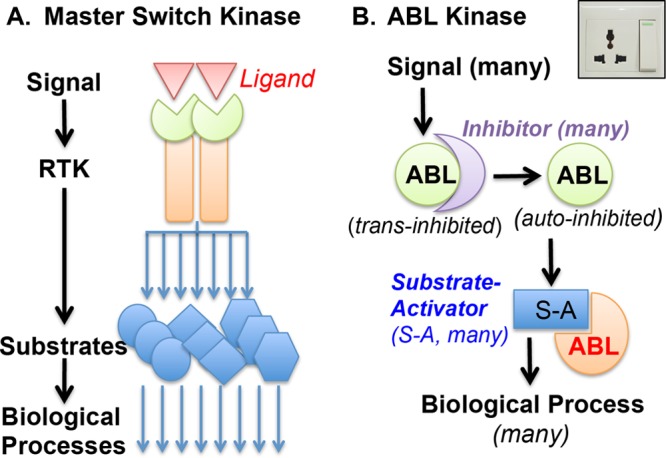
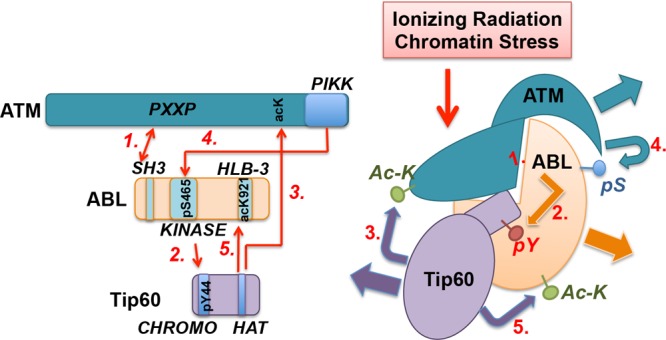
The mammalian ABL1 gene encodes the ubiquitously expressed nonreceptor tyrosine kinase ABL. In response to growth factors, cytokines, cell adhesion, DNA damage, oxidative stress, and other signals, ABL is activated to stimulate cell proliferation or differentiation, survival or death, retraction, or migration. ABL also regulates specialized functions such as antigen receptor signaling in lymphocytes, synapse formation in neurons, and bacterial adhesion to intestinal epithelial cells. Although discovered as the proto-oncogene from which the Abelson leukemia virus derived its Gag-v-Abl oncogene, recent results have linked ABL kinase activation to neuronal degeneration. This body of knowledge on ABL seems confusing because it does not fit the one-gene-one-function paradigm. Without question, ABL capabilities are encoded by its gene sequence and that molecular blueprint designs this kinase to be regulated by subcellular location-dependent interactions with inhibitors and substrate activators. Furthermore, ABL shuttles between the nucleus and the cytoplasm where it binds DNA and actin--two biopolymers with fundamental roles in almost all biological processes. Taken together, the cumulated results from analyses of ABL structure-function, ABL mutant mouse phenotypes, and ABL substrates suggest that this tyrosine kinase does not have its own agenda but that, instead, it has evolved to serve a variety of tissue-specific and context-dependent biological functions.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous