

A p38MAPK/MK2 signaling pathway leading to redox stress, cell death and ischemia/reperfusion injury
- PMID: 24423080
- PMCID: PMC3896752
- DOI: 10.1186/1478-811X-12-6
A p38MAPK/MK2 signaling pathway leading to redox stress, cell death and ischemia/reperfusion injury
Abstract
Background: Many diseases and pathological conditions are characterized by transient or constitutive overproduction of reactive oxygen species (ROS). ROS are causal for ischemia/reperfusion (IR)-associated tissue injury (IRI), a major contributor to organ dysfunction or failure. Preventing IRI with antioxidants failed in the clinic, most likely due to the difficulty to timely and efficiently target them to the site of ROS production and action. IR is also characterized by changes in the activity of intracellular signaling molecules including the stress kinase p38MAPK. While ROS can cause the activation of p38MAPK, we recently obtained in vitro evidence that p38MAPK activation is responsible for elevated mitochondrial ROS levels, thus suggesting a role for p38MAPK upstream of ROS and their damaging effects.
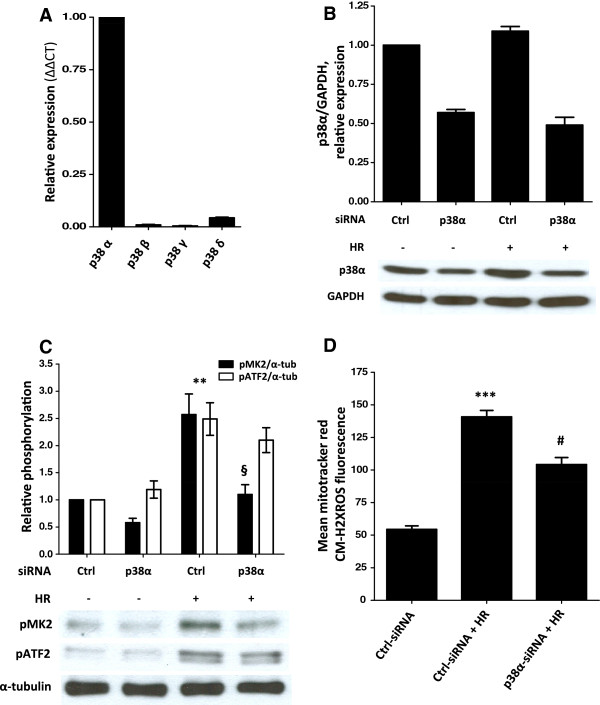
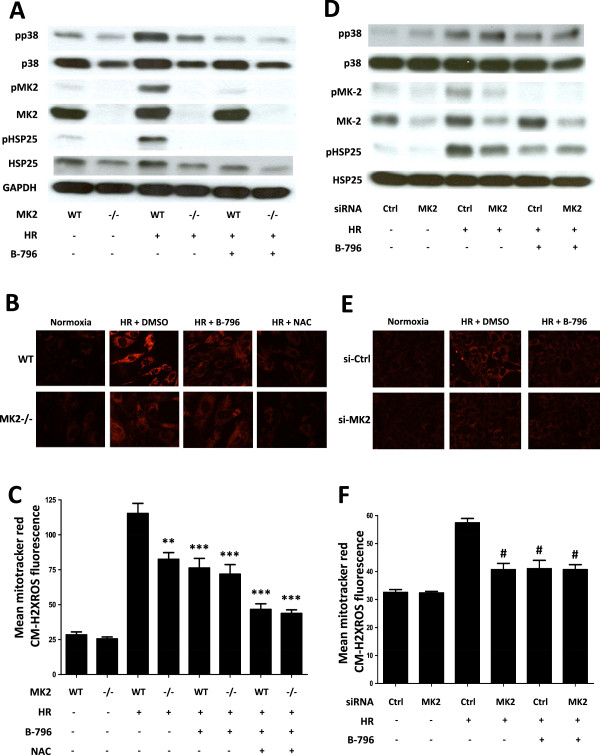
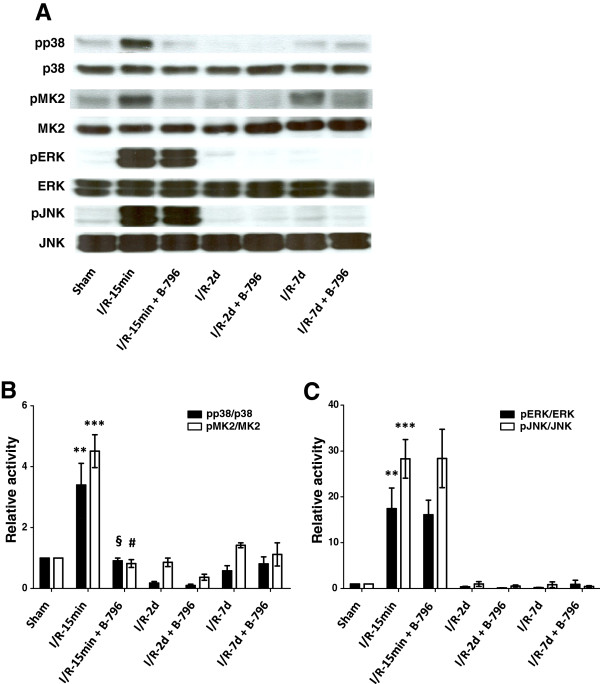
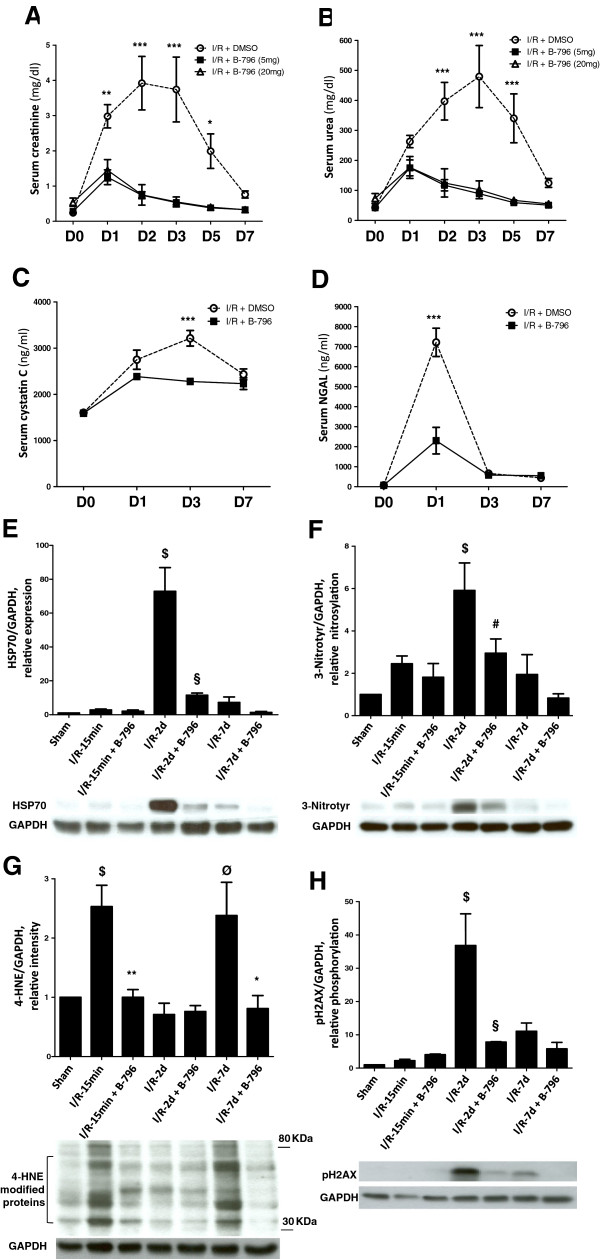
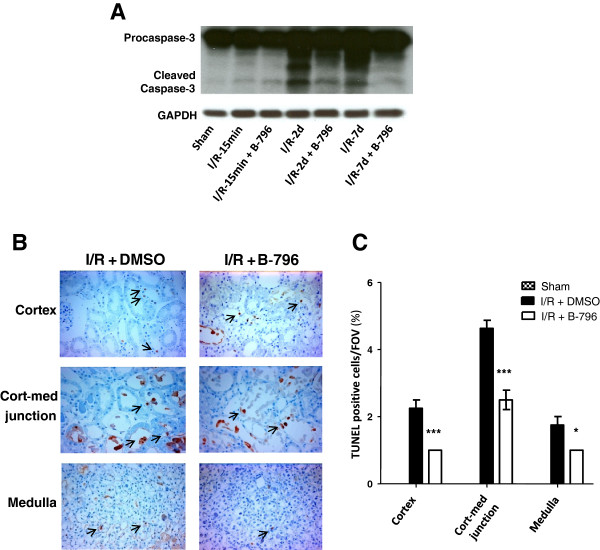
Results: Here we identified p38MAPKα as the predominantly expressed isoform in HL-1 cardiomyocytes and siRNA-mediated knockdown demonstrated the pro-oxidant role of p38MAPKα signaling. Moreover, the knockout of the p38MAPK effector MAPKAP kinase 2 (MK2) reproduced the effect of inhibiting or knocking down p38MAPK. To translate these findings into a setting closer to the clinic a stringent kidney clamping model was used. p38MAPK activity increased upon reperfusion and p38MAPK inhibition by the inhibitor BIRB796 almost completely prevented severe functional impairment caused by IR. Histological and molecular analyses showed that protection resulted from decreased redox stress and apoptotic cell death.
Conclusions: These data highlight a novel and important mechanism for p38MAPK to cause IRI and suggest it as a potential therapeutic target for prevention of tissue injury.
Figures
Similar articles
-
MK2-/- gene knockout mouse hearts carry anti-apoptotic signal and are resistant to ischemia reperfusion injury.J Mol Cell Cardiol. 2005 Jan;38(1):93-7. doi: 10.1016/j.yjmcc.2004.10.018. Epub 2004 Dec 9. J Mol Cell Cardiol. 2005. PMID: 15623425
-
DUSP14 knockout accelerates cardiac ischemia reperfusion (IR) injury through activating NF-κB and MAPKs signaling pathways modulated by ROS generation.Biochem Biophys Res Commun. 2018 Jun 18;501(1):24-32. doi: 10.1016/j.bbrc.2018.04.101. Epub 2018 May 8. Biochem Biophys Res Commun. 2018. PMID: 29660332
-
Apelin protects against ischemia-reperfusion injury in diabetic myocardium via inhibiting apoptosis and oxidative stress through PI3K and p38-MAPK signaling pathways.Aging (Albany NY). 2020 Dec 20;12(24):25120-25137. doi: 10.18632/aging.104106. Epub 2020 Dec 20. Aging (Albany NY). 2020. PMID: 33342766 Free PMC article.
-
Protecting the heart through MK2 modulation, toward a role in diabetic cardiomyopathy and lipid metabolism.Biochim Biophys Acta Mol Basis Dis. 2018 May;1864(5 Pt B):1914-1922. doi: 10.1016/j.bbadis.2017.07.015. Epub 2017 Jul 19. Biochim Biophys Acta Mol Basis Dis. 2018. PMID: 28735097 Review.
-
Novel Therapeutic Potential of Mitogen-Activated Protein Kinase Activated Protein Kinase 2 (MK2) in Chronic Airway Inflammatory Disorders.Curr Drug Targets. 2019;20(4):367-379. doi: 10.2174/1389450119666180816121323. Curr Drug Targets. 2019. PMID: 30112991 Review.
Cited by
-
An optimized low-pressure tourniquet murine hind limb ischemia reperfusion model: Inducing acute ischemia reperfusion injury in C57BL/6 wild type mice.PLoS One. 2019 Jan 24;14(1):e0210961. doi: 10.1371/journal.pone.0210961. eCollection 2019. PLoS One. 2019. PMID: 30677066 Free PMC article.
-
Dioscin Attenuates Myocardial Ischemic/Reperfusion-Induced Cardiac Dysfunction through Suppression of Reactive Oxygen Species.Oxid Med Cell Longev. 2021 Oct 5;2021:3766919. doi: 10.1155/2021/3766919. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34664015 Free PMC article.
-
Role of Oxidative Stress in Reperfusion following Myocardial Ischemia and Its Treatments.Oxid Med Cell Longev. 2021 May 18;2021:6614009. doi: 10.1155/2021/6614009. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34055195 Free PMC article. Review.
-
Protective Effects of Nuciferine in Middle Cerebral Artery Occlusion Rats Based on Transcriptomics.Brain Sci. 2022 Apr 28;12(5):572. doi: 10.3390/brainsci12050572. Brain Sci. 2022. PMID: 35624959 Free PMC article.
-
Biochanin A Alleviates Cerebral Ischemia/Reperfusion Injury by Suppressing Endoplasmic Reticulum Stress-Induced Apoptosis and p38MAPK Signaling Pathway In Vivo and In Vitro.Front Endocrinol (Lausanne). 2021 Jul 12;12:646720. doi: 10.3389/fendo.2021.646720. eCollection 2021. Front Endocrinol (Lausanne). 2021. PMID: 34322090 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
