

Delineation of C12orf65-related phenotypes: a genotype-phenotype relationship
- PMID: 24424123
- PMCID: PMC4350599
- DOI: 10.1038/ejhg.2013.284
Delineation of C12orf65-related phenotypes: a genotype-phenotype relationship
Abstract
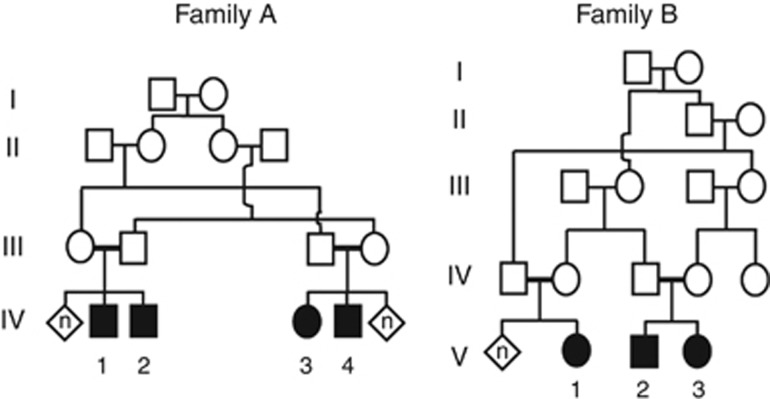
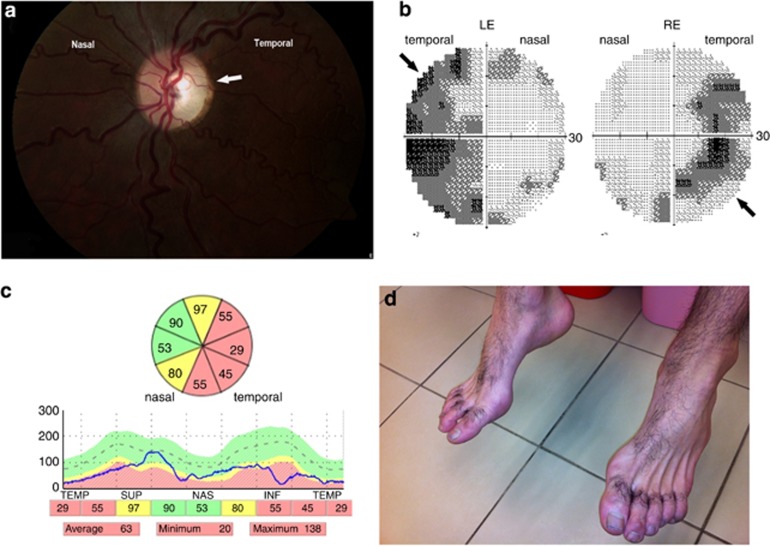
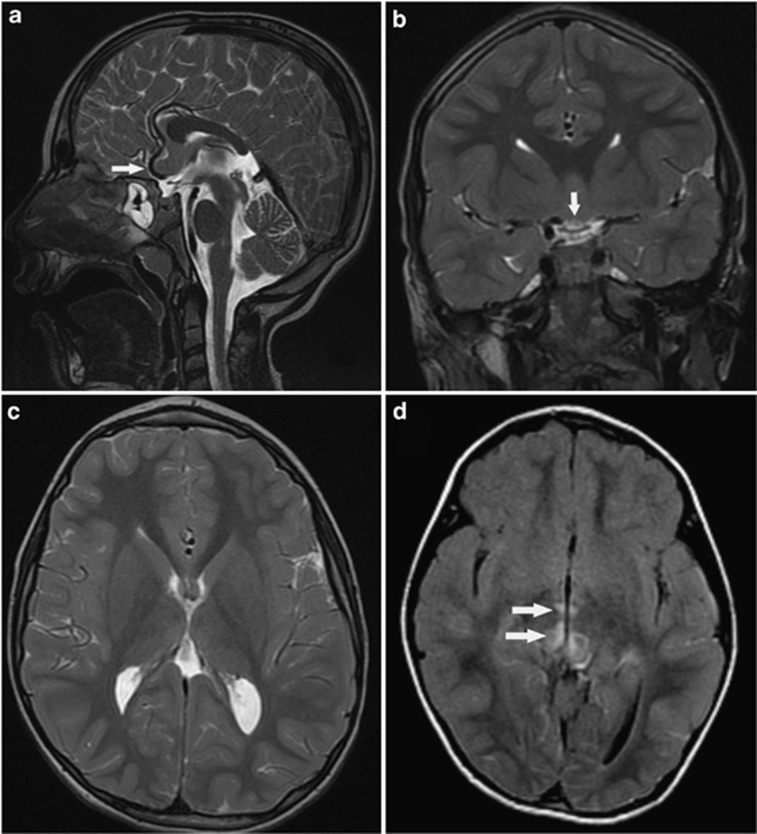
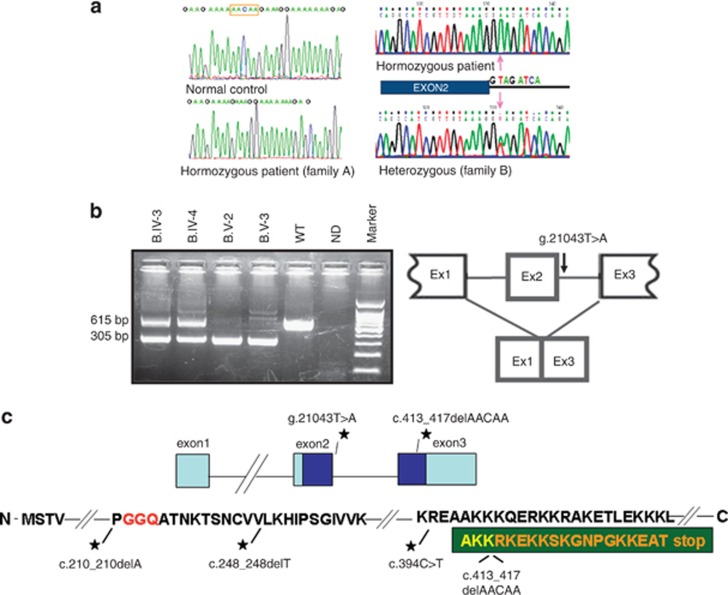
C12orf65 participates in the process of mitochondrial translation and has been shown to be associated with a spectrum of phenotypes, including early onset optic atrophy, progressive encephalomyopathy, peripheral neuropathy, and spastic paraparesis.We used whole-genome homozygosity mapping as well as exome sequencing and targeted gene sequencing to identify novel C12orf65 disease-causing mutations in seven affected individuals originating from two consanguineous families. In four family members affected with childhood-onset optic atrophy accompanied by slowly progressive peripheral neuropathy and spastic paraparesis, we identified a homozygous frame shift mutation c.413_417 delAACAA, which predicts a truncated protein lacking the C-terminal portion. In the second family, we studied three affected individuals who presented with early onset optic atrophy, peripheral neuropathy, and spastic gait in addition to moderate intellectual disability. Muscle biopsy in two of the patients revealed decreased activities of the mitochondrial respiratory chain complexes I and IV. In these patients, we identified a homozygous splice mutation, g.21043 T>A (c.282+2 T>A) which leads to skipping of exon 2. Our study broadens the phenotypic spectrum of C12orf65 defects and highlights the triad of optic atrophy, axonal neuropathy and spastic paraparesis as its key clinical features. In addition, a clear genotype-phenotype correlation is anticipated in which deleterious mutations which disrupt the GGQ-containing domain in the first coding exon are expected to result in a more severe phenotype, whereas down-stream C-terminal mutations may result in a more favorable phenotype, typically lacking cognitive impairment.
Figures
Comment in
-
Applications of next-generation whole exome sequencing.J Neurol. 2014 Jun;261(6):1244-6. doi: 10.1007/s00415-014-7372-1. J Neurol. 2014. PMID: 24838538 No abstract available.
References
-
- Lightowlers RN, Chrzanowska-Lightowlers ZM. Terminating human mitochondrial protein synthesis: a shift in our thinking. RNA Biol. 2010;7:282–286. - PubMed
-
- Shimazaki H, Takiyama Y, Ishiura H, et al. A homozygous mutation of C12orf65 causes spastic paraplegia with optic atrophy and neuropathy (SPG55) J Med Genet. 2012;49:777–784. - PubMed
-
- Saada A, Bar-Meir M, Belaiche C, Miller C, Elpeleg O. Evaluation of enzymatic assays and compounds affecting ATP production in mitochondrial respiratory chain complex I deficiency. Anal Biochem. 2004;335:66–72. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
