

Deep sequencing of evolving pathogen populations: applications, errors, and bioinformatic solutions
- PMID: 24428920
- PMCID: PMC3902414
- DOI: 10.1186/2042-5783-4-1
Deep sequencing of evolving pathogen populations: applications, errors, and bioinformatic solutions
Abstract
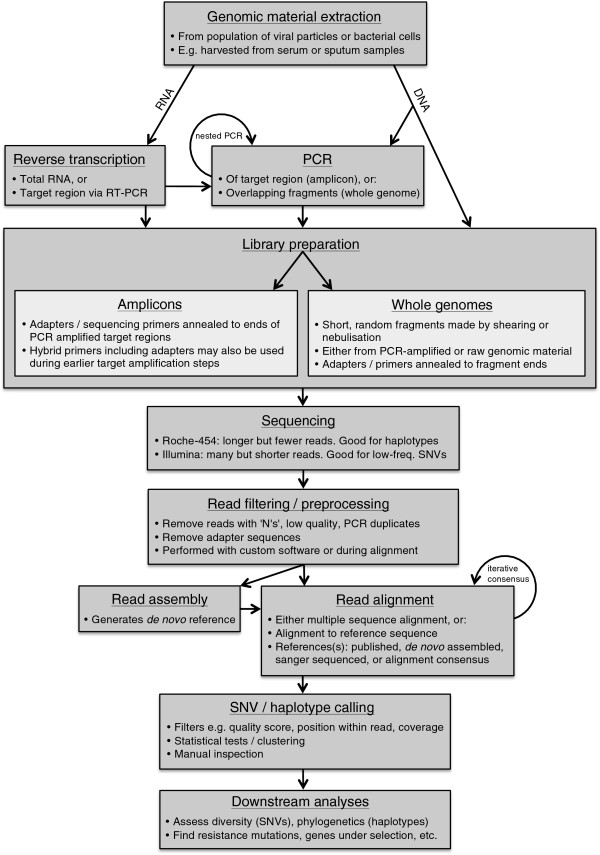
Deep sequencing harnesses the high throughput nature of next generation sequencing technologies to generate population samples, treating information contained in individual reads as meaningful. Here, we review applications of deep sequencing to pathogen evolution. Pioneering deep sequencing studies from the virology literature are discussed, such as whole genome Roche-454 sequencing analyses of the dynamics of the rapidly mutating pathogens hepatitis C virus and HIV. Extension of the deep sequencing approach to bacterial populations is then discussed, including the impacts of emerging sequencing technologies. While it is clear that deep sequencing has unprecedented potential for assessing the genetic structure and evolutionary history of pathogen populations, bioinformatic challenges remain. We summarise current approaches to overcoming these challenges, in particular methods for detecting low frequency variants in the context of sequencing error and reconstructing individual haplotypes from short reads.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources