

Heterogeneity of genomic evolution and mutational profiles in multiple myeloma
- PMID: 24429703
- PMCID: PMC3905727
- DOI: 10.1038/ncomms3997
Heterogeneity of genomic evolution and mutational profiles in multiple myeloma
Abstract
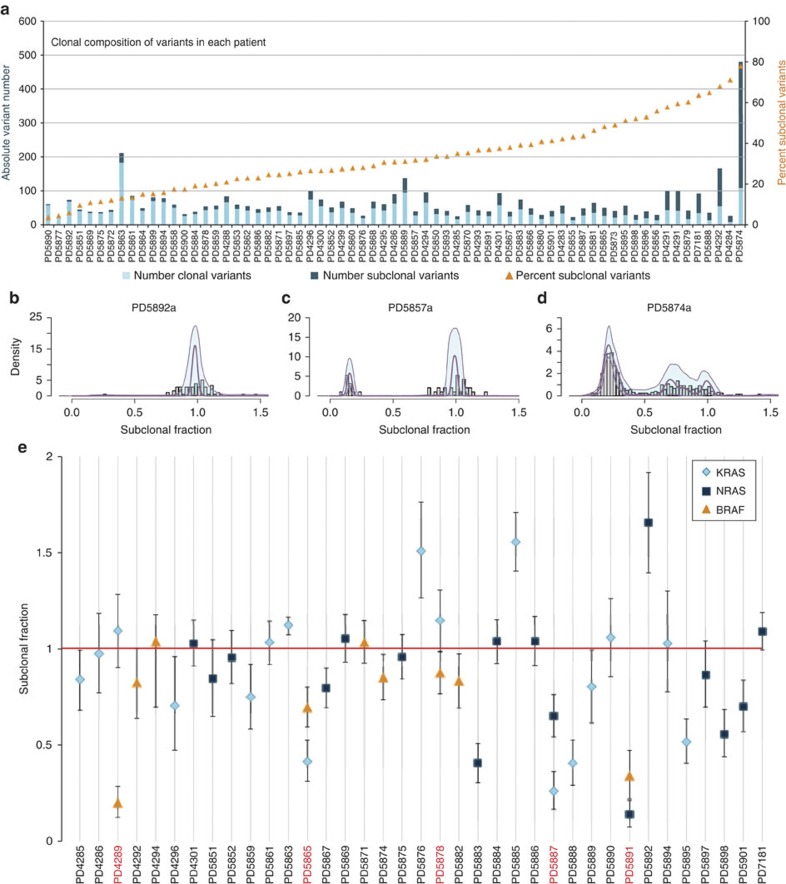
Multiple myeloma is an incurable plasma cell malignancy with a complex and incompletely understood molecular pathogenesis. Here we use whole-exome sequencing, copy-number profiling and cytogenetics to analyse 84 myeloma samples. Most cases have a complex subclonal structure and show clusters of subclonal variants, including subclonal driver mutations. Serial sampling reveals diverse patterns of clonal evolution, including linear evolution, differential clonal response and branching evolution. Diverse processes contribute to the mutational repertoire, including kataegis and somatic hypermutation, and their relative contribution changes over time. We find heterogeneity of mutational spectrum across samples, with few recurrent genes. We identify new candidate genes, including truncations of SP140, LTB, ROBO1 and clustered missense mutations in EGR1. The myeloma genome is heterogeneous across the cohort, and exhibits diversity in clonal admixture and in dynamics of evolution, which may impact prognostic stratification, therapeutic approaches and assessment of disease response to treatment.
Figures





References
-
- Bergsagel P. L. & Kuehl W. M. Molecular pathogenesis and a consequent classification of multiple myeloma. J. Clin. Oncol. 23, 6333–6338 (2005). - PubMed
-
- Hideshima T., Mitsiades C., Tonon G., Richardson P. G. & Anderson K. C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer. 7, 585–598 (2007). - PubMed
-
- Morgan G. J., Walker B. A. & Davies F. E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer. 12, 335–348 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01-78378/PHS HHS/United States
- R01 CA125711/CA/NCI NIH HHS/United States
- R01-124929/PHS HHS/United States
- P01 CA078378/CA/NCI NIH HHS/United States
- P01 CA155258/CA/NCI NIH HHS/United States
- 077012/Z/05/Z/WT_/Wellcome Trust/United Kingdom
- P50 CA100707/CA/NCI NIH HHS/United States
- T32 CA106209/CA/NCI NIH HHS/United States
- P50-100007/PHS HHS/United States
- P01-155258/PHS HHS/United States
- I01 BX001584/BX/BLRD VA/United States
- RCA125711C/PHS HHS/United States
- 088340/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
