

A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome
- PMID: 24431331
- PMCID: PMC4278941
- DOI: 10.1136/jmedgenet-2013-101660
A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome
Abstract
Introduction: Lenz microphthalmia syndrome (LMS) is a genetically heterogeneous X-linked disorder characterised by microphthalmia/anophthalmia, skeletal abnormalities, genitourinary malformations, and anomalies of the digits, ears, and teeth. Intellectual disability and seizure disorders are seen in about 60% of affected males. To date, no gene has been identified for LMS in the microphthalmia syndrome 1 locus (MCOPS1). In this study, we aim to find the disease-causing gene for this condition.
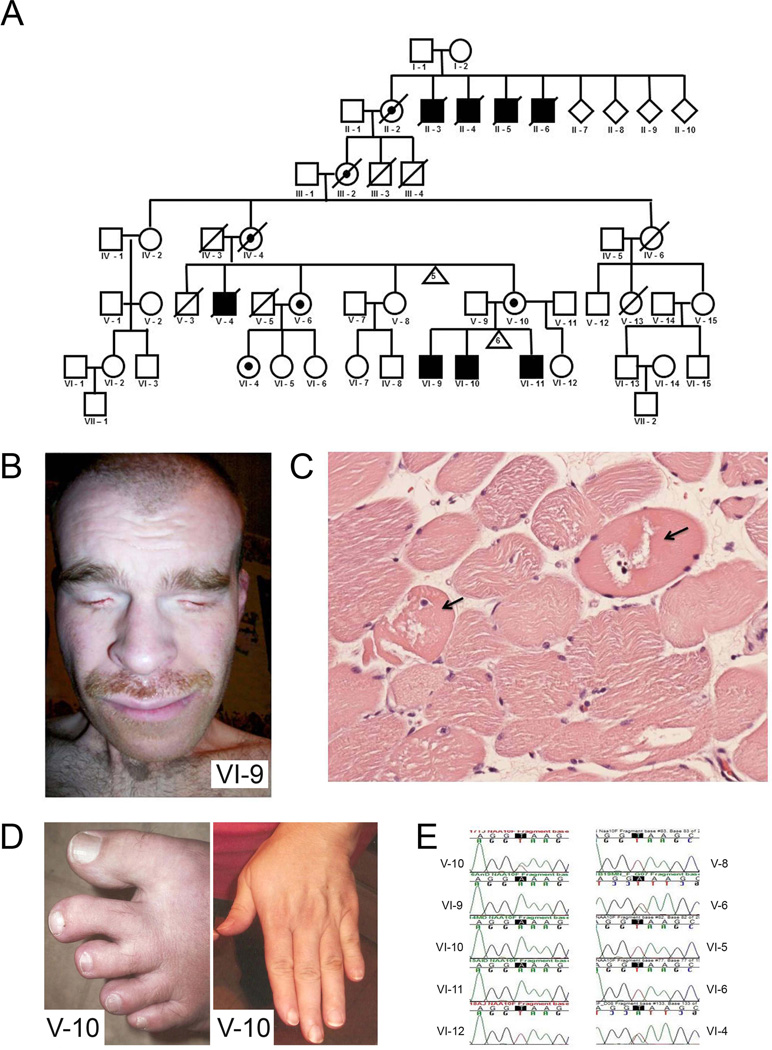
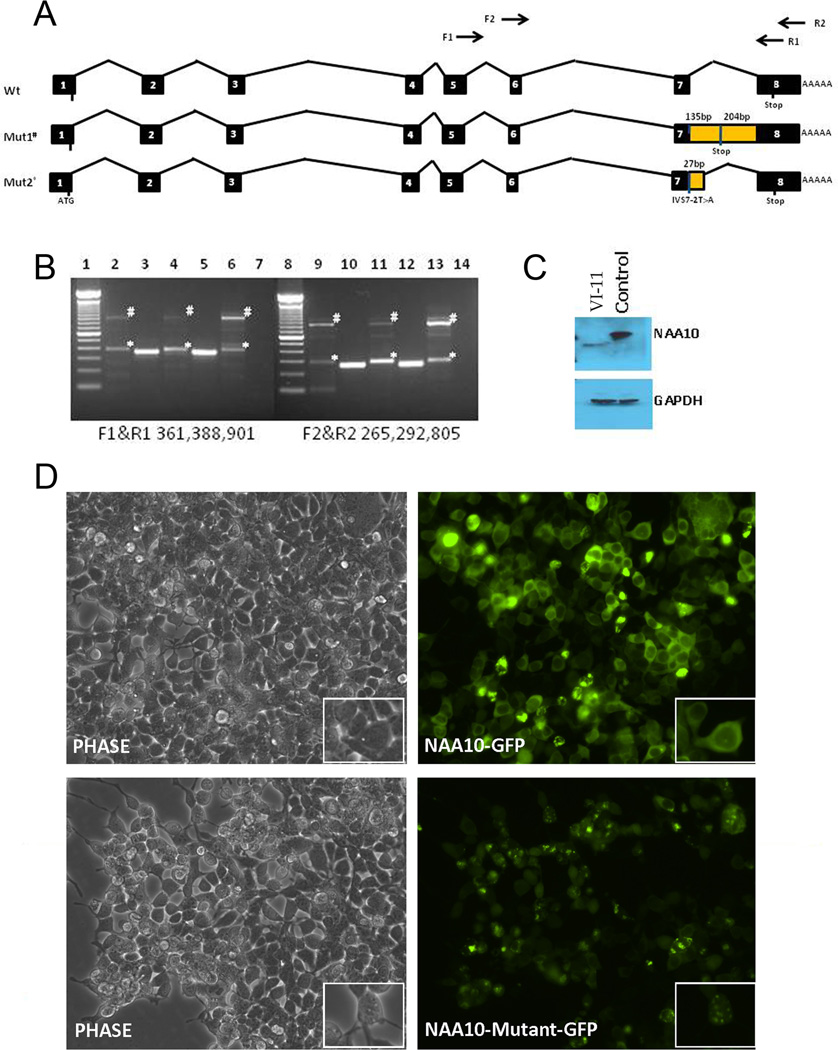
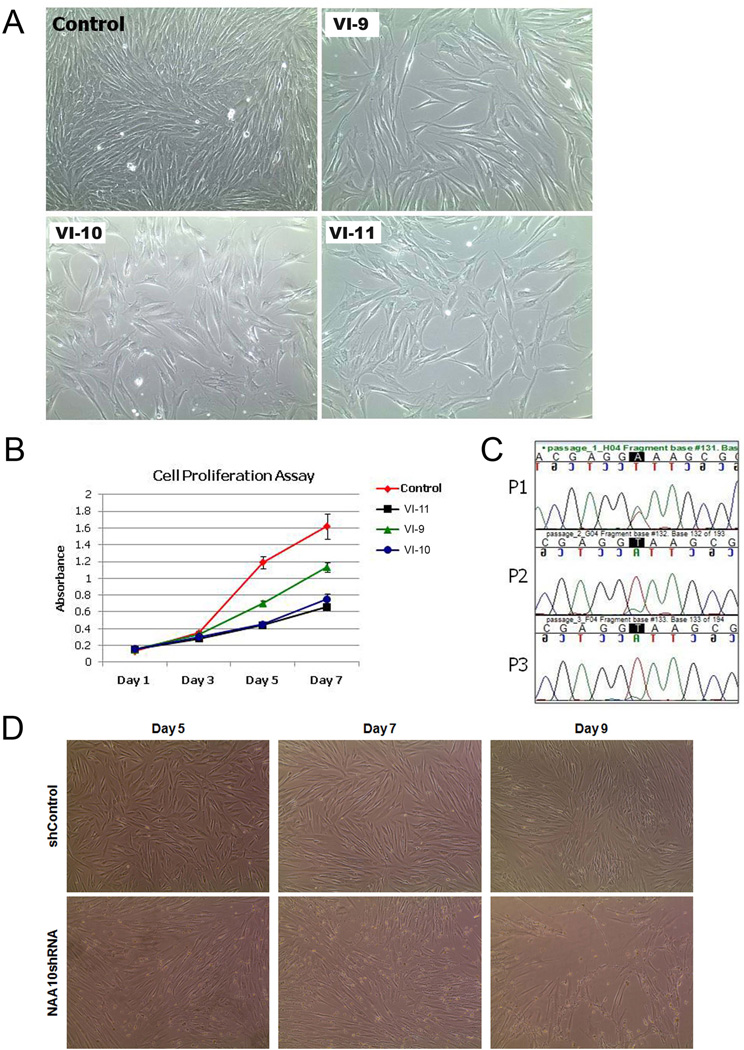
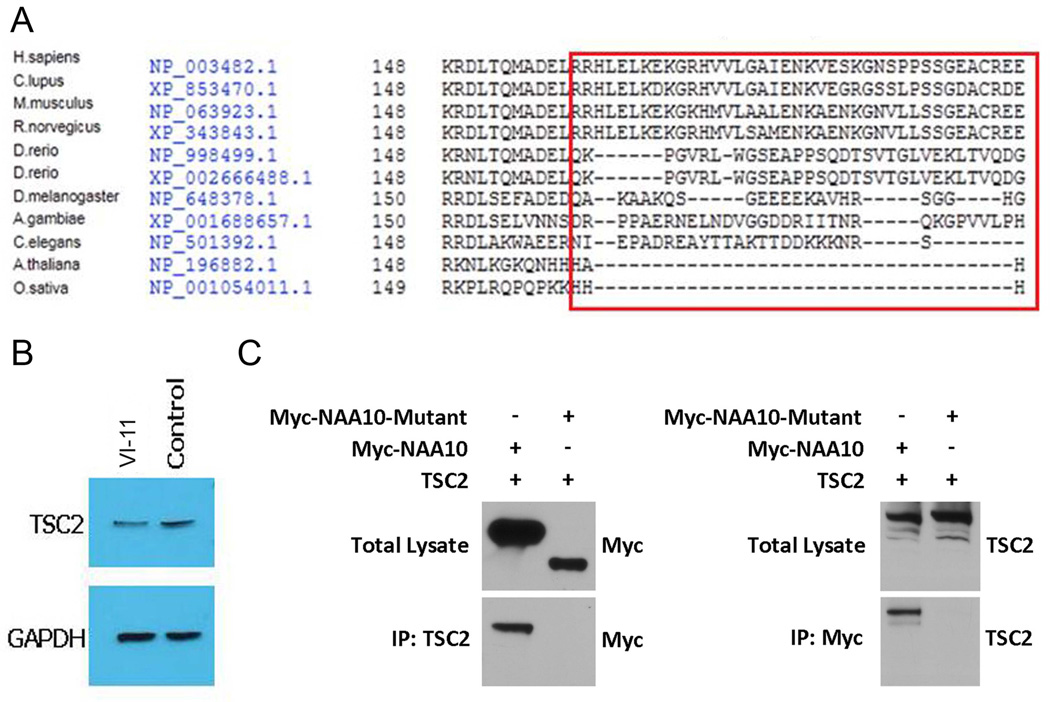
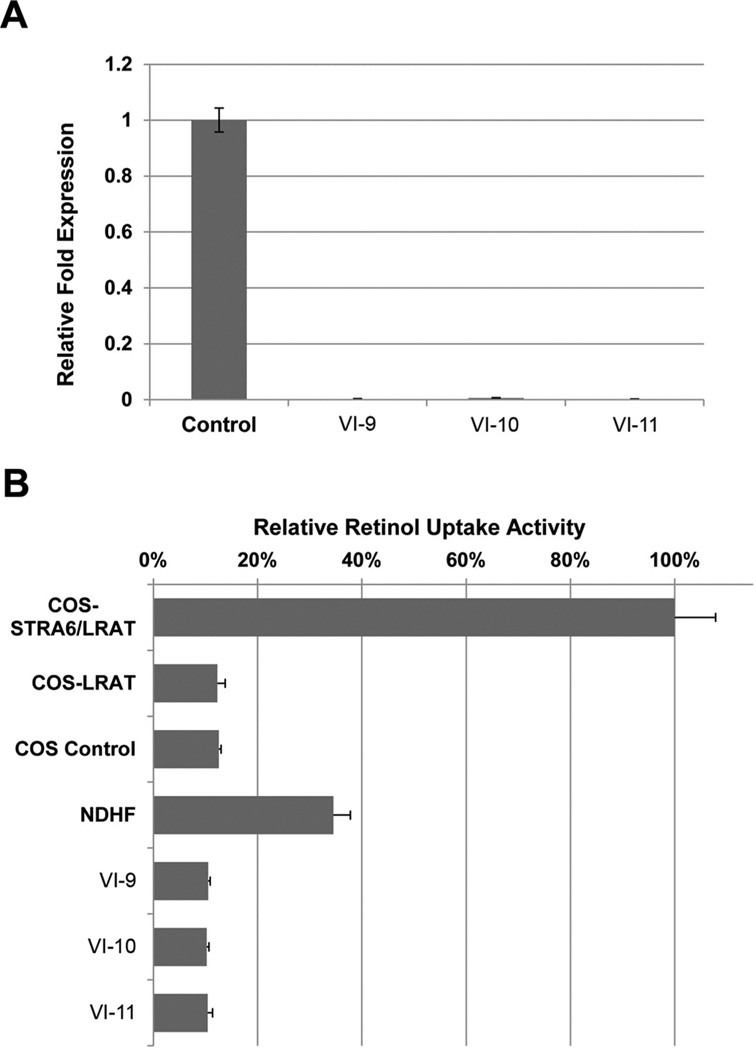
Methods and results: Using exome sequencing in a family with three affected brothers, we identified a mutation in the intron 7 splice donor site (c.471+2T→A) of the N-acetyltransferase NAA10 gene. NAA10 has been previously shown to be mutated in patients with Ogden syndrome, which is clinically distinct from LMS. Linkage studies for this family mapped the disease locus to Xq27-Xq28, which was consistent with the locus of NAA10. The mutation co-segregated with the phenotype and cDNA analysis showed aberrant transcripts. Patient fibroblasts lacked expression of full length NAA10 protein and displayed cell proliferation defects. Expression array studies showed significant dysregulation of genes associated with genetic forms of anophthalmia such as BMP4, STRA6, and downstream targets of BCOR and the canonical WNT pathway. In particular, STRA6 is a retinol binding protein receptor that mediates cellular uptake of retinol/vitamin A and plays a major role in regulating the retinoic acid signalling pathway. A retinol uptake assay showed that retinol uptake was decreased in patient cells.
Conclusions: We conclude that the NAA10 mutation is the cause of LMS in this family, likely through the dysregulation of the retinoic acid signalling pathway.
Keywords: Clinical Genetics; Developmental; Genome-Wide; Lenz Microphthalmia Syndrome; NAA10.
Figures
References
-
- Traboulsi EI, Lenz W, Gonzales-Ramos M, Siegel J, Macrae WG, Maumenee IH. The Lenz microphthalmia syndrome. Am J Ophthalmol. 1988;105:40–45. - PubMed
-
- Graham CA, Redmond RM, Nevin NC. X-linked clinical anophthalmos. Localization of the gene to Xq27-Xq28. Ophthalmic Paediatr Genet. 1991;12:43–48. - PubMed
-
- Forrester S, Kovach MJ, Reynolds NM, Urban R, Kimonis V. Manifestations in four males with and an obligate carrier of the Lenz microphthalmia syndrome. Am J Med Genet. 2001;98:92–100. - PubMed
-
- Ng D, Hadley DW, Tifft CJ, Biesecker LG. Genetic heterogeneity of syndromic X-linked recessive microphthalmia-anophthalmia: is Lenz microphthalmia a single disorder? Am J Med Genet. 2002;110:308–314. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases