

Molecular mechanisms of hepatic apoptosis
- PMID: 24434519
- PMCID: PMC4040708
- DOI: 10.1038/cddis.2013.499
Molecular mechanisms of hepatic apoptosis
Abstract
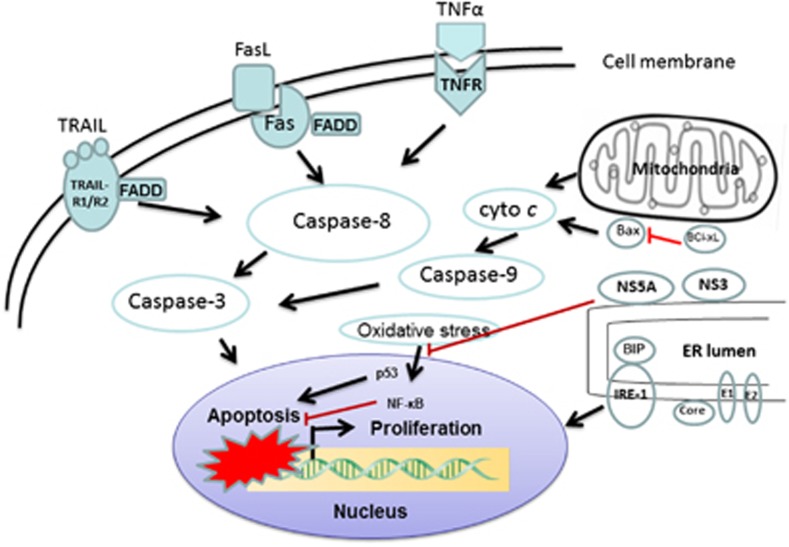
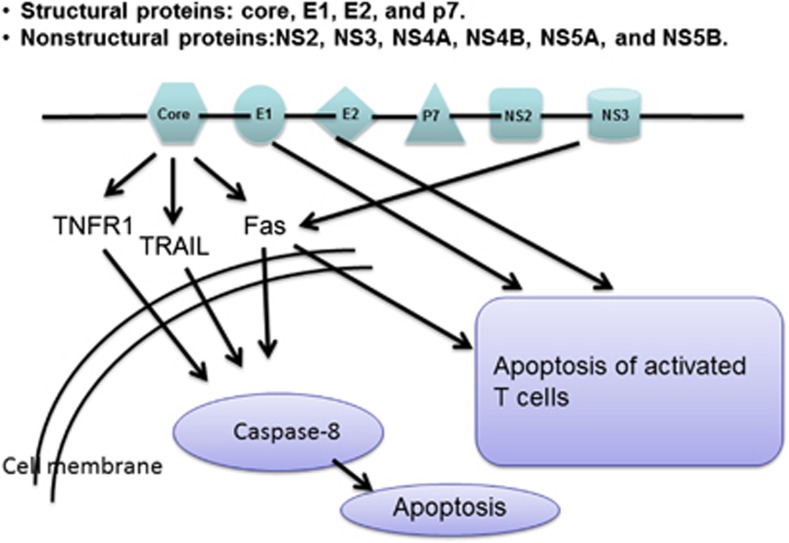
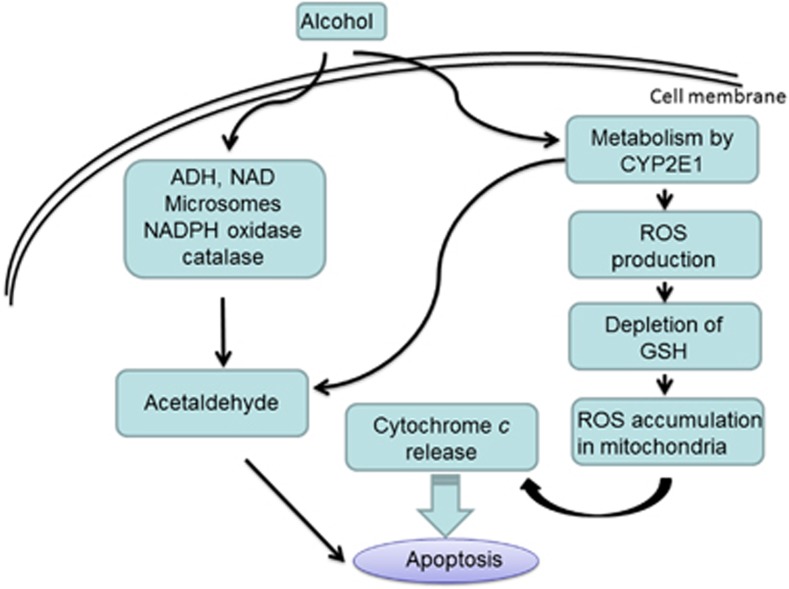
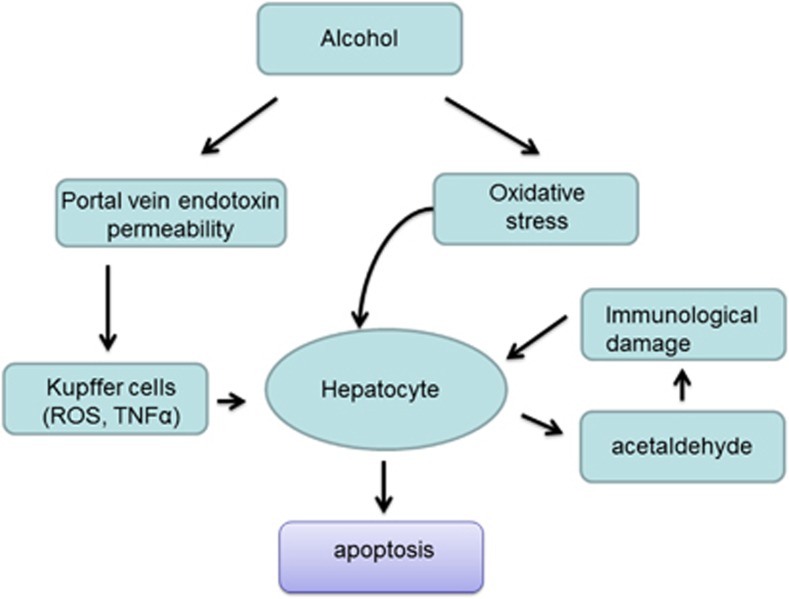
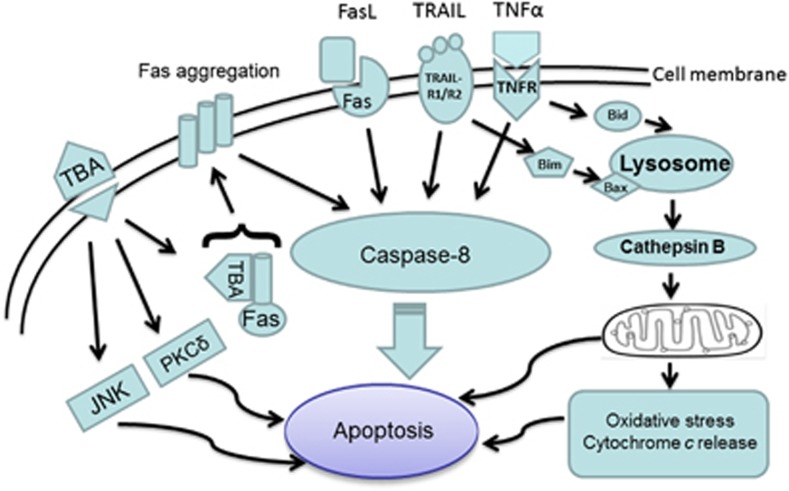
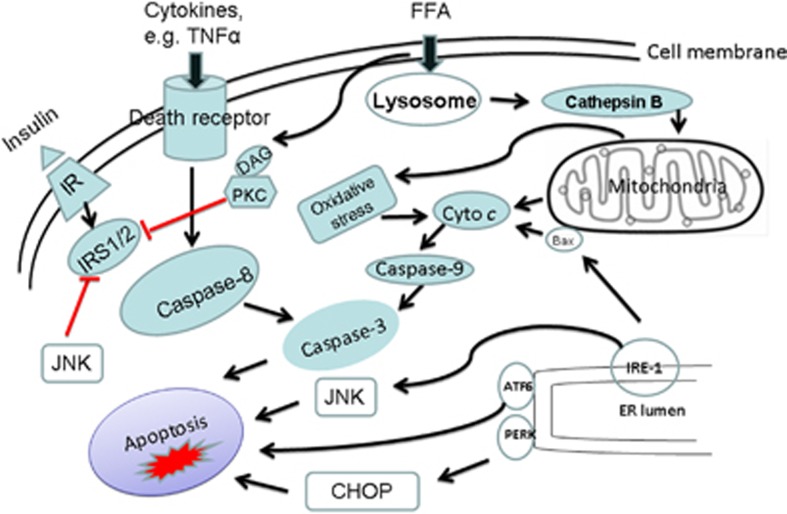
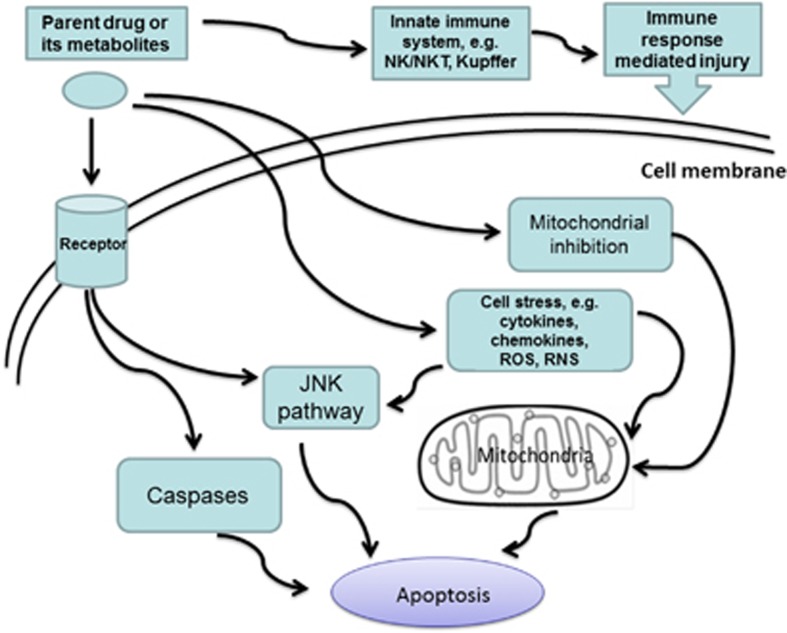
Apoptosis is a prominent feature of liver diseases. Causative factors such as alcohol, viruses, toxic bile acids, fatty acids, drugs, and immune response, can induce apoptotic cell death via membrane receptors and intracellular stress. Apoptotic signaling network, including membrane death receptor-mediated cascade, reactive oxygen species (ROS) generation, endoplasmic reticulum (ER) stress, lysosomal permeabilization, and mitochondrial dysfunction, is intermixed each other, but one mechanism may dominate at a particular stage. Mechanisms of hepatic apoptosis are complicated by multiple signaling pathways. The progression of liver disease is affected by the balance between apoptotic and antiapoptotic capabilities. Therapeutic options of liver injury are impacted by the clear understanding toward mechanisms of hepatic apoptosis.
Figures

References
-
- Smith PG, Tee LB, Yeoh GC. Appearance of oval cells in the liver of rats after long-term exposure to ethanol. Hepatology. 1996;23:145–154. - PubMed
-
- Sun C, Jin XL, Xiao JC. Oval cells in hepatitis B virus-positive and hepatitis C virus-positive liver cirrhosis: histological and ultrastructural study. Histopathology. 2006;48:546–555. - PubMed
-
- Soden JS, Devereaux MW, Haas JE, Gumpricht E, Dahl R, Gralla J, et al. Subcutaneous vitamin E ameliorates liver injury in an in vivo model of steatocholestasis. Hepatology. 2007;46:485–495. - PubMed
-
- Dahl TB, Haukeland JW, Yndestad A, Ranheim T, Gladhaug IP, Damas JK, et al. Intracellular nicotinamide phosphoribosyltransferase protects against hepatocyte apoptosis and is down-regulated in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2010;95:3039–3047. - PubMed
-
- Waldhauser KM, Torok M, Ha HR, Thomet U, Konrad D, Brecht K, et al. Hepatocellular toxicity and pharmacological effect of amiodarone and amiodarone derivatives. J Pharmacol Exp Ther. 2006;319:1413–1423. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
