

Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling
- PMID: 24436048
- PMCID: PMC4049524
- DOI: 10.1158/2159-8290.CD-13-0611
Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling
Abstract
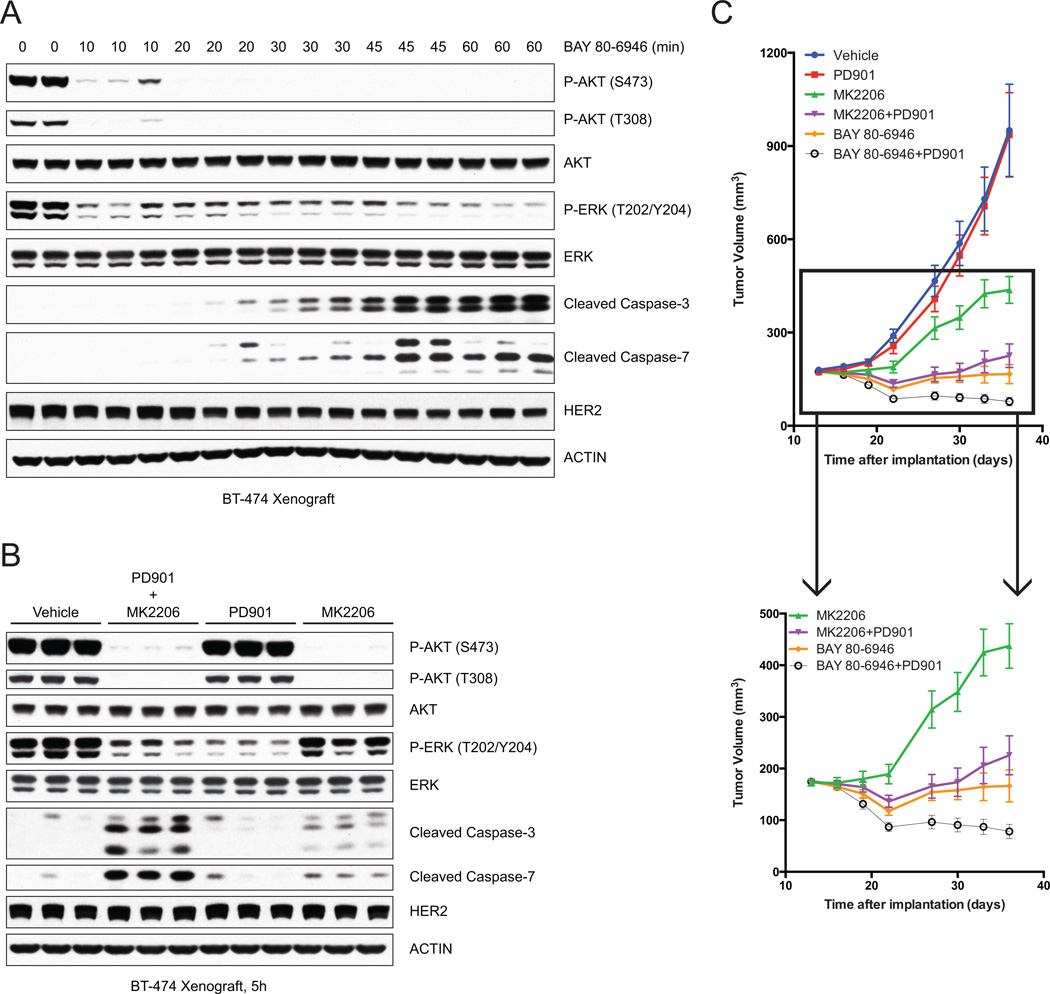
The effects of selective phosphoinositide 3-kinase (PI3K) and AKT inhibitors were compared in human tumor cell lines in which the pathway is dysregulated. Both caused inhibition of AKT, relief of feedback inhibition of receptor tyrosine kinases, and growth arrest. However, only the PI3K inhibitors caused rapid induction of cell death. In seeking a mechanism for this phenomenon, we found that PI3K inhibition, but not AKT inhibition, causes rapid inhibition of wild-type RAS and of RAF-MEK-ERK signaling. Inhibition of RAS-ERK signaling is transient, rebounding a few hours after drug addition, and is required for rapid induction of apoptosis. Combined MEK and AKT inhibition also promotes cell death, and in murine models of HER2(+) cancer, either pulsatile PI3K inhibition or combined MEK and AKT inhibition causes tumor regression. We conclude that PI3K is upstream of RAS and AKT and that pulsatile inhibition of both pathways is sufficient for effective antitumor activity.
Conflict of interest statement
Claudia Schneider and Ningshu Liu are employees of Bayer Research Labs
No other author has a conflict of interest to disclose.
Figures






Comment in
-
Signalling: a clearer pathway view.Nat Rev Cancer. 2014 Mar;14(3):156-7. doi: 10.1038/nrc3682. Epub 2014 Feb 6. Nat Rev Cancer. 2014. PMID: 24500377 No abstract available.
-
Vascular disease: a new way to starve vascular endothelial cells.Nat Rev Drug Discov. 2014 Mar;13(3):176-7. doi: 10.1038/nrd4264. Epub 2014 Feb 14. Nat Rev Drug Discov. 2014. PMID: 24525783 No abstract available.
References
-
- Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. - PubMed
-
- Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer research. 2005;65:2554–2559. - PubMed
-
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. - PubMed
-
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
