

A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington's disease
- PMID: 24436303
- PMCID: PMC4031626
- DOI: 10.1093/hmg/ddu010
A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington's disease
Abstract
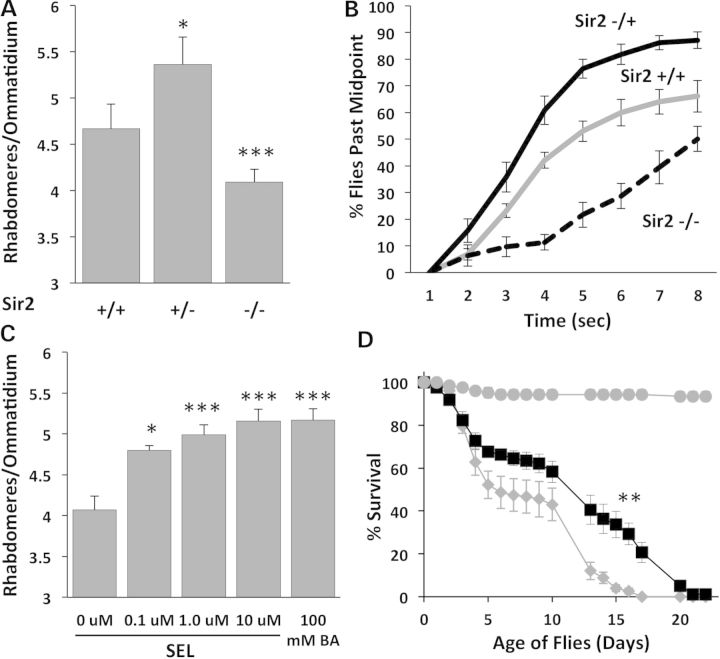
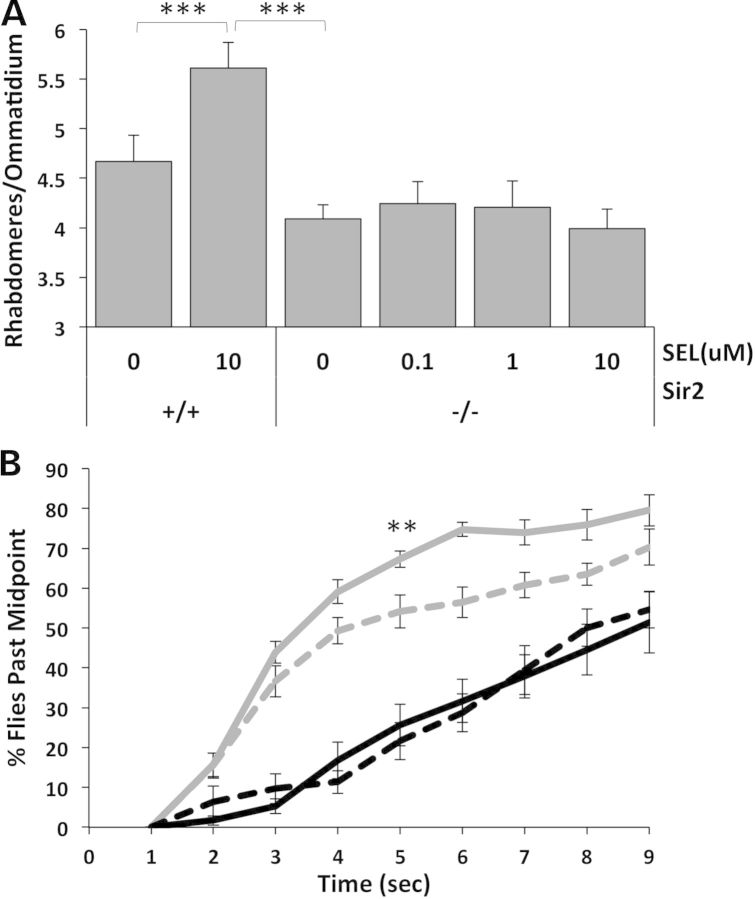
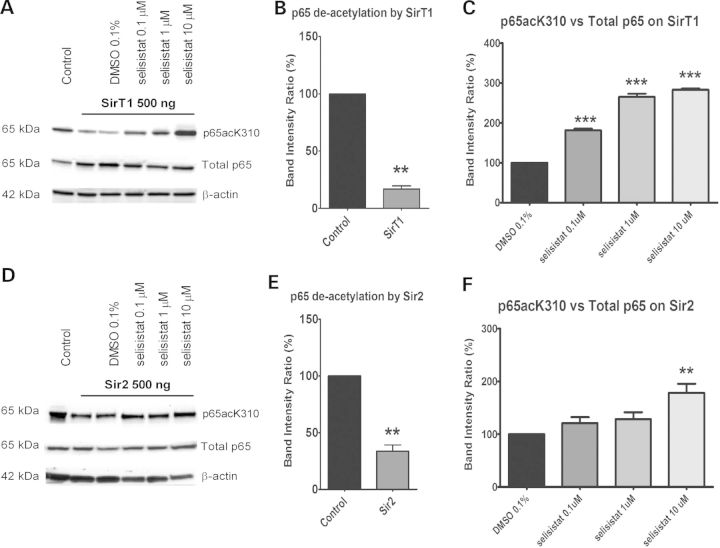
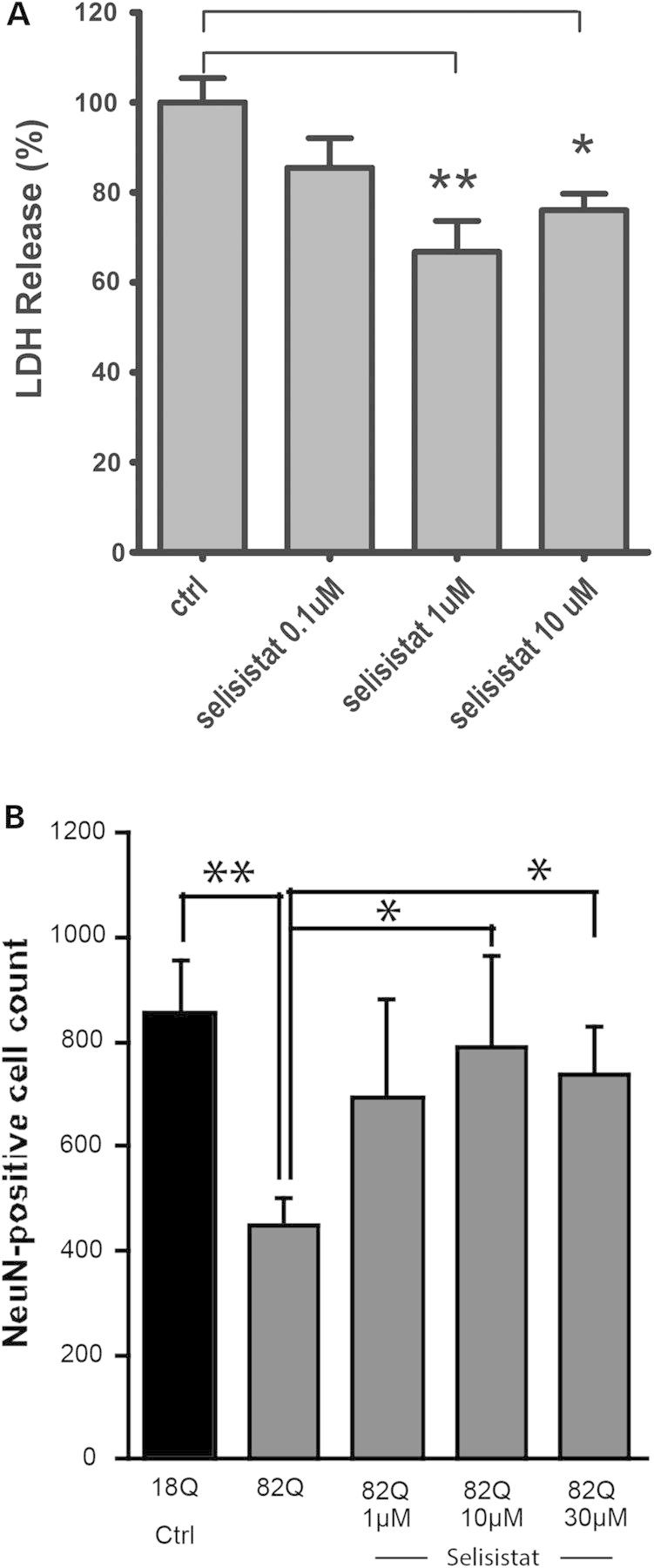
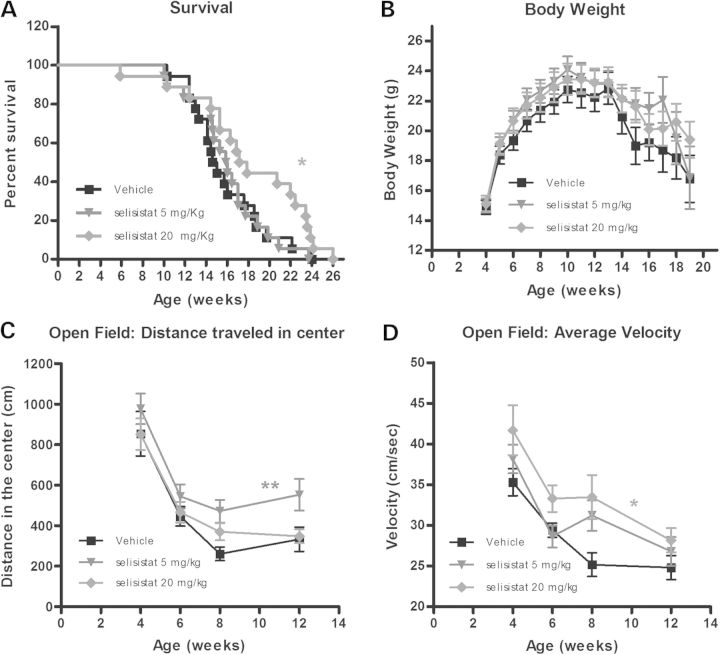
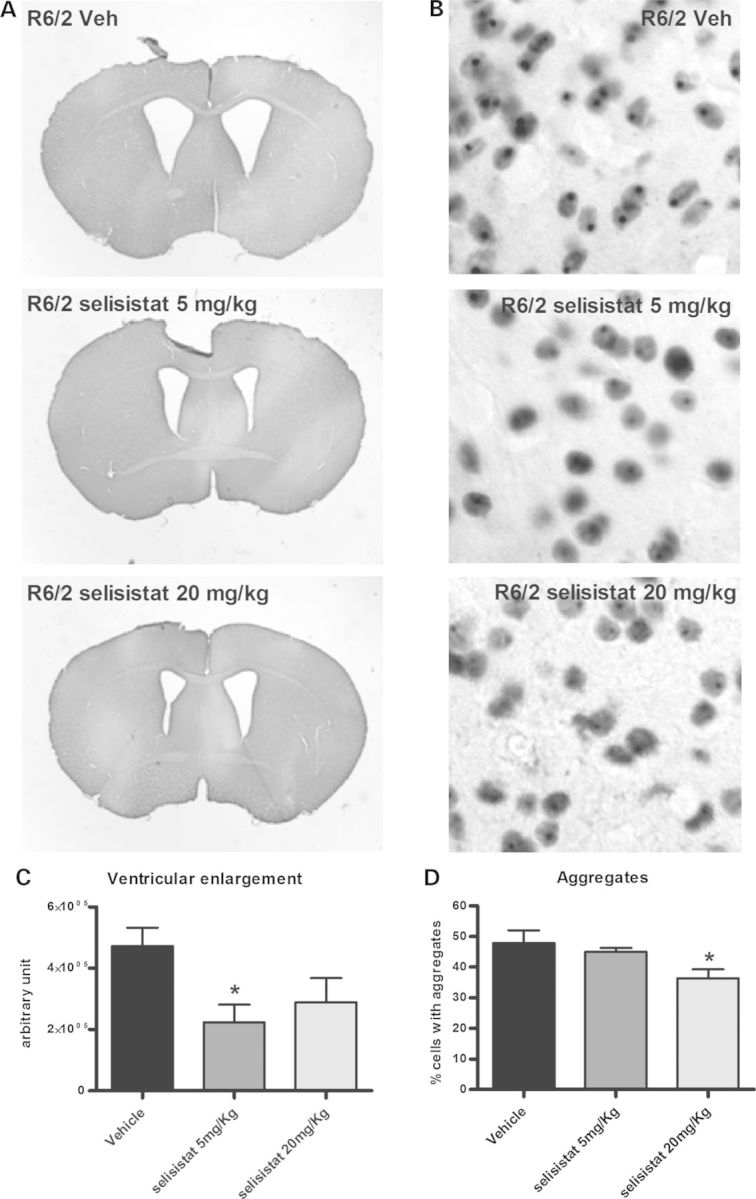
Protein acetylation, which is central to transcriptional control as well as other cellular processes, is disrupted in Huntington's disease (HD). Treatments that restore global acetylation levels, such as inhibiting histone deacetylases (HDACs), are effective in suppressing HD pathology in model organisms. However, agents that selectively target the disease-relevant HDACs have not been available. SirT1 (Sir2 in Drosophila melanogaster) deacetylates histones and other proteins including transcription factors. Genetically reducing, but not eliminating, Sir2 has been shown to suppress HD pathology in model organisms. To date, small molecule inhibitors of sirtuins have exhibited low potency and unattractive pharmacological and biopharmaceutical properties. Here, we show that highly selective pharmacological inhibition of Drosophila Sir2 and mammalian SirT1 using the novel inhibitor selisistat (selisistat; 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide) can suppress HD pathology caused by mutant huntingtin exon 1 fragments in Drosophila, mammalian cells and mice. We have validated Sir2 as the in vivo target of selisistat by showing that genetic elimination of Sir2 eradicates the effect of this inhibitor in Drosophila. The specificity of selisistat is shown by its effect on recombinant sirtuins in mammalian cells. Reduction of HD pathology by selisistat in Drosophila, mammalian cells and mouse models of HD suggests that this inhibitor has potential as an effective therapeutic treatment for human disease and may also serve as a tool to better understand the downstream pathways of SirT1/Sir2 that may be critical for HD.
Figures
Similar articles
-
Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington's disease.Hum Mol Genet. 2008 Dec 1;17(23):3767-75. doi: 10.1093/hmg/ddn273. Epub 2008 Sep 1. Hum Mol Genet. 2008. PMID: 18762557 Free PMC article.
-
Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington's disease.Neurobiol Dis. 2012 May;46(2):351-61. doi: 10.1016/j.nbd.2012.01.016. Neurobiol Dis. 2012. PMID: 22590724 Free PMC article.
-
An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington's disease.Br J Clin Pharmacol. 2015 Mar;79(3):465-76. doi: 10.1111/bcp.12512. Br J Clin Pharmacol. 2015. PMID: 25223731 Free PMC article. Clinical Trial.
-
Sirtuins as Modifiers of Huntington's Disease (HD) Pathology.Prog Mol Biol Transl Sci. 2018;154:105-145. doi: 10.1016/bs.pmbts.2017.11.013. Epub 2017 Dec 27. Prog Mol Biol Transl Sci. 2018. PMID: 29413175 Review.
-
The interaction between FOXO and SIRT1: tipping the balance towards survival.Trends Cell Biol. 2004 Aug;14(8):408-12. doi: 10.1016/j.tcb.2004.07.006. Trends Cell Biol. 2004. PMID: 15308206 Review.
Cited by
-
Synaptic pathology in Huntington's disease: Beyond the corticostriatal pathway.Neurobiol Dis. 2022 Jan;162:105574. doi: 10.1016/j.nbd.2021.105574. Epub 2021 Nov 27. Neurobiol Dis. 2022. PMID: 34848336 Free PMC article. Review.
-
SIRT1 and SIRT2 Activity Control in Neurodegenerative Diseases.Front Pharmacol. 2021 Jan 12;11:585821. doi: 10.3389/fphar.2020.585821. eCollection 2020. Front Pharmacol. 2021. PMID: 33597872 Free PMC article. Review.
-
SIRT1 Activity Is Linked to Its Brain Region-Specific Phosphorylation and Is Impaired in Huntington's Disease Mice.PLoS One. 2016 Jan 27;11(1):e0145425. doi: 10.1371/journal.pone.0145425. eCollection 2016. PLoS One. 2016. PMID: 26815359 Free PMC article.
-
SIRT1 in the brain-connections with aging-associated disorders and lifespan.Front Cell Neurosci. 2015 Mar 9;9:64. doi: 10.3389/fncel.2015.00064. eCollection 2015. Front Cell Neurosci. 2015. PMID: 25805970 Free PMC article. Review.
-
Molecular Imaging of Sirtuin1 Expression-Activity in Rat Brain Using Positron-Emission Tomography-Magnetic-Resonance Imaging with [18F]-2-Fluorobenzoylaminohexanoicanilide.J Med Chem. 2018 Aug 23;61(16):7116-7130. doi: 10.1021/acs.jmedchem.8b00253. Epub 2018 Aug 13. J Med Chem. 2018. PMID: 30052441 Free PMC article.
References
-
- Cha J.H. Transcriptional dysregulation in Huntington's disease. Trends Neurosci. 2000;23:387–392. - PubMed
-
- Ferrante R.J., Kubilus J.K., Lee J., Ryu H., Beesen A., Zucker B., Smith K., Kowall N.W., Ratan R.R., Luthi-Carter R., et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J. Neurosci. 2003;23:9418–9427. - PMC - PubMed
-
- Sadri-Vakili G., Cha J.H. Mechanisms of disease: Histone modifications in Huntington's disease. Nat. Clin. Pract. Neurol. 2006;2: 330–338. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
