

Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation
- PMID: 24439110
- PMCID: PMC3928656
- DOI: 10.1016/j.ajhg.2013.12.012
Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation
Abstract
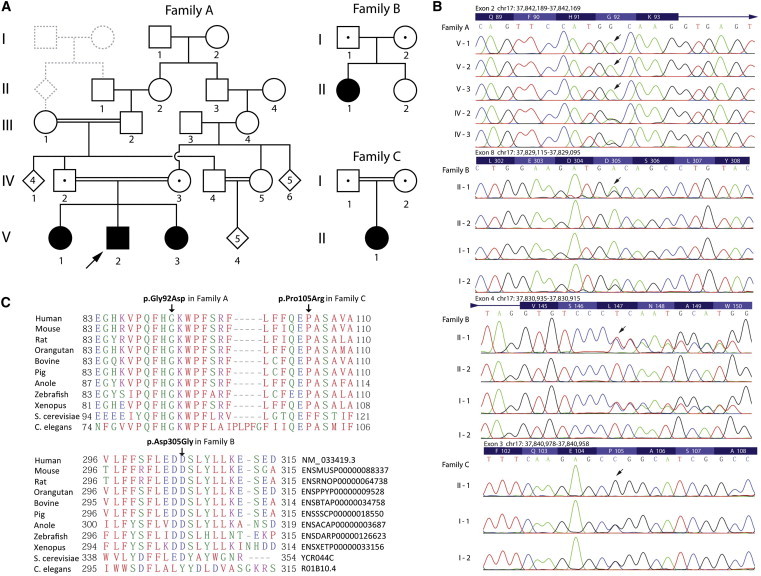
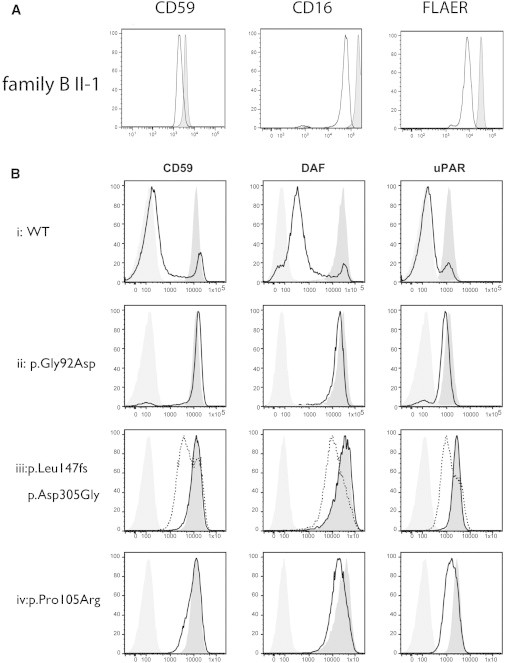
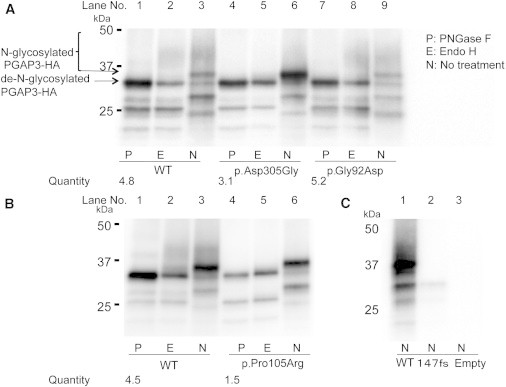
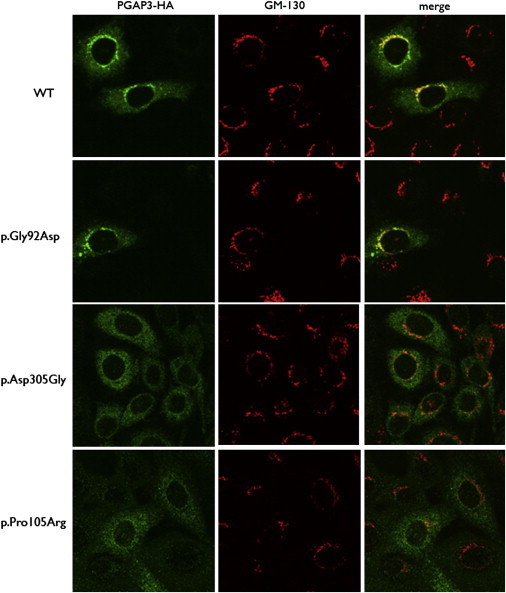
Glycosylphophatidylinositol (GPI)-anchored proteins play important roles in many biological processes, and mutations affecting proteins involved in the synthesis of the GPI anchor are reported to cause a wide spectrum of intellectual disabilities (IDs) with characteristic additional phenotypic features. Here, we describe a total of five individuals (from three unrelated families) in whom we identified mutations in PGAP3, encoding a protein that is involved in GPI-anchor maturation. Three siblings in a consanguineous Pakistani family presented with profound developmental delay, severe ID, no speech, psychomotor delay, and postnatal microcephaly. A combination of autozygosity mapping and exome sequencing identified a 13.8 Mb region harboring a homozygous c.275G>A (p.Gly92Asp) variant in PGAP3 region 17q11.2-q21.32. Subsequent testing showed elevated serum alkaline phosphatase (ALP), a GPI-anchored enzyme, in all three affected children. In two unrelated individuals in a cohort with developmental delay, ID, and elevated ALP, we identified compound-heterozygous variants c.439dupC (p.Leu147Profs(∗)16) and c.914A>G (p.Asp305Gly) and homozygous variant c.314C>G (p.Pro105Arg). The 1 bp duplication causes a frameshift and nonsense-mediated decay. Further evidence supporting pathogenicity of the missense mutations c.275G>A, c.314C>G, and c.914A>G was provided by the absence of the variants from ethnically matched controls, phylogenetic conservation, and functional studies on Chinese hamster ovary cell lines. Taken together with recent data on PGAP2, these results confirm the importance of the later GPI-anchor remodelling steps for normal neuronal development. Impairment of PGAP3 causes a subtype of hyperphosphatasia with ID, a congenital disorder of glycosylation that is also referred to as Mabry syndrome.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Fujita M., Kinoshita T. GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim. Biophys. Acta. 2012;1821:1050–1058. - PubMed
-
- Jaeken J. Congenital disorders of glycosylation (CDG): it’s (nearly) all in it! J. Inherit. Metab. Dis. 2011;34:853–858. - PubMed
-
- Nozaki M., Ohishi K., Yamada N., Kinoshita T., Nagy A., Takeda J. Developmental abnormalities of glycosylphosphatidylinositol-anchor-deficient embryos revealed by Cre/loxP system. Lab. Invest. 1999;79:293–299. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
