

Improvement of experimental testing and network training conditions with genome-wide microarrays for more accurate predictions of drug gene targets
- PMID: 24444313
- PMCID: PMC3911882
- DOI: 10.1186/1752-0509-8-7
Improvement of experimental testing and network training conditions with genome-wide microarrays for more accurate predictions of drug gene targets
Abstract
Background: Genome-wide microarrays have been useful for predicting chemical-genetic interactions at the gene level. However, interpreting genome-wide microarray results can be overwhelming due to the vast output of gene expression data combined with off-target transcriptional responses many times induced by a drug treatment. This study demonstrates how experimental and computational methods can interact with each other, to arrive at more accurate predictions of drug-induced perturbations. We present a two-stage strategy that links microarray experimental testing and network training conditions to predict gene perturbations for a drug with a known mechanism of action in a well-studied organism.
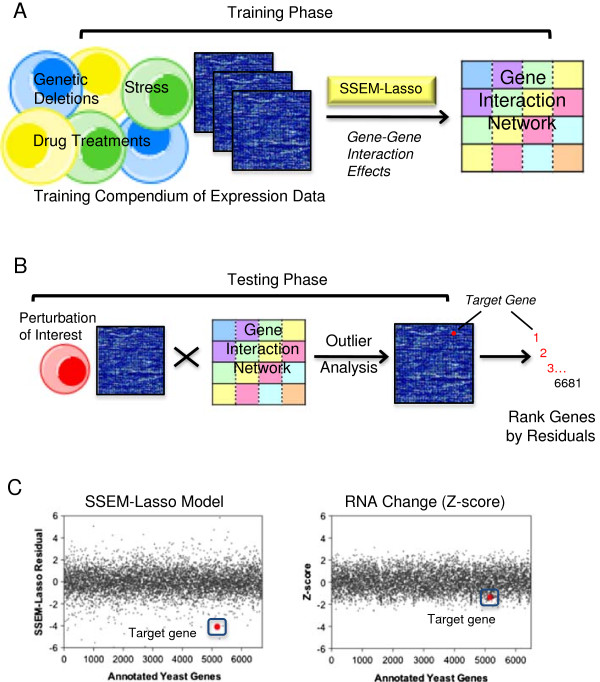
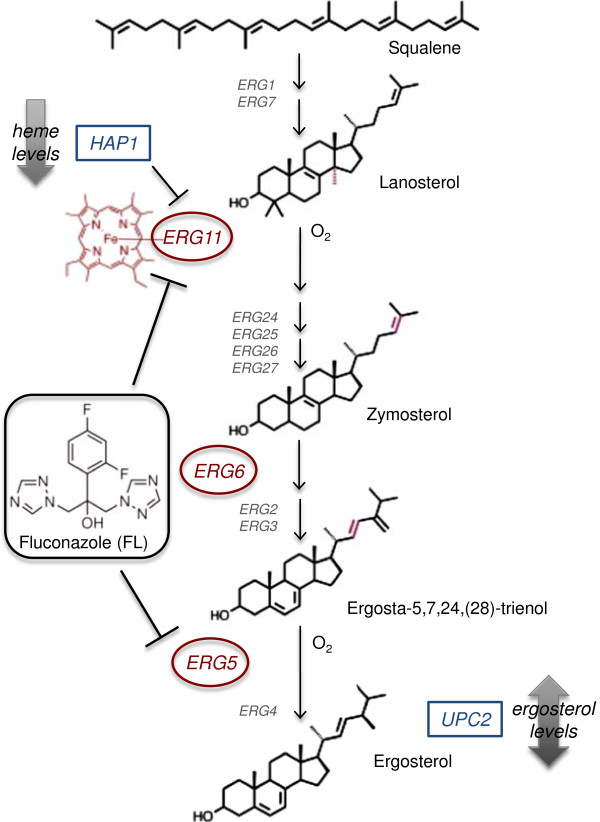
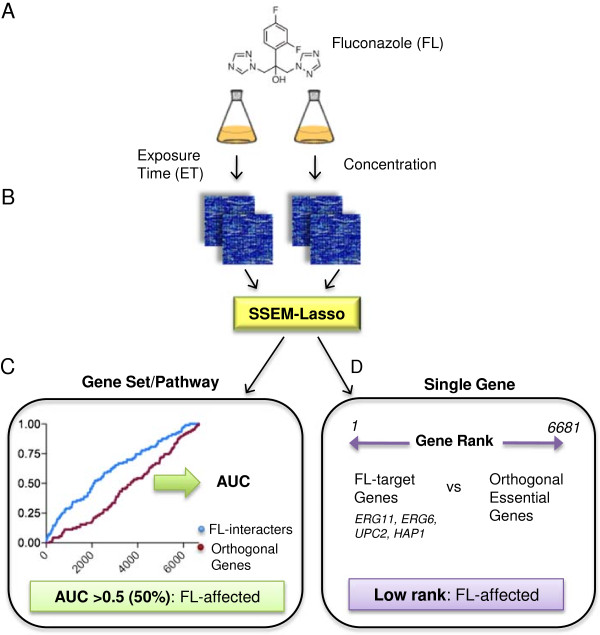
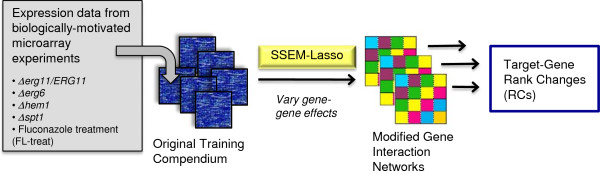
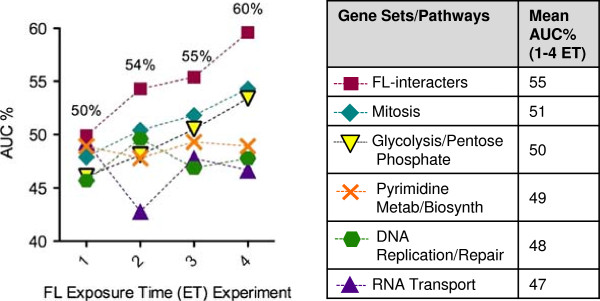
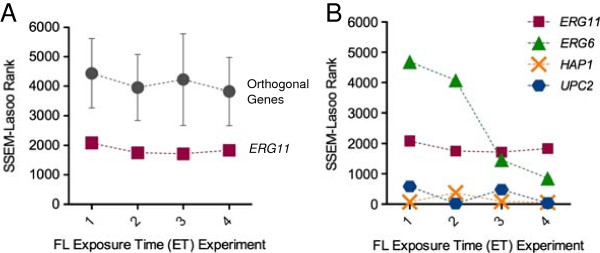
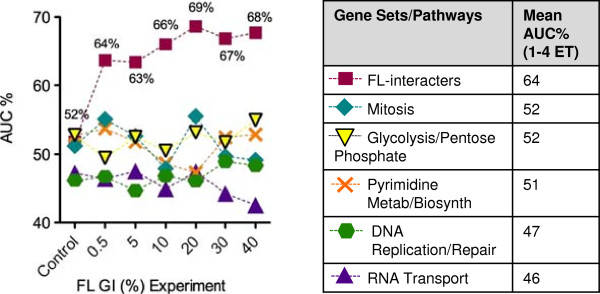
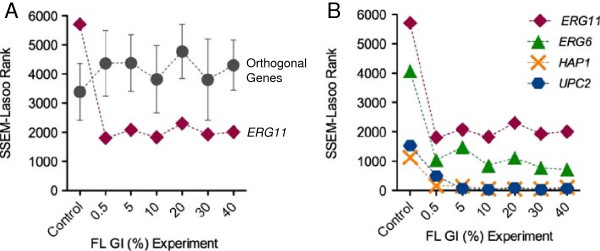
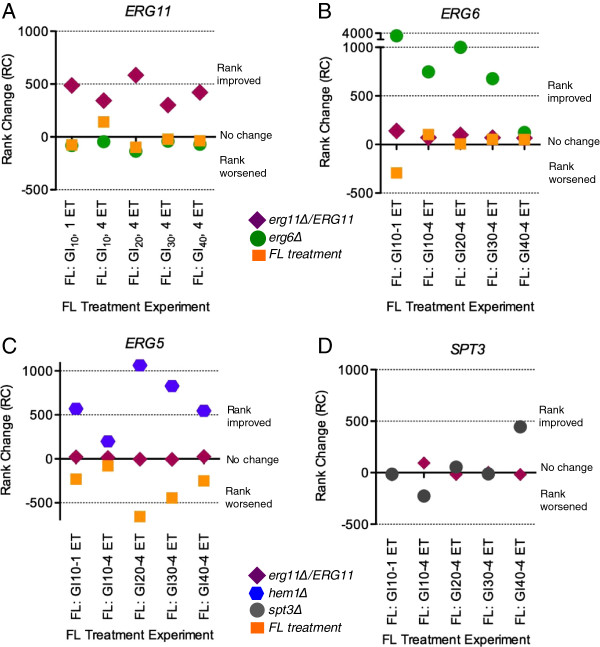
Results: S. cerevisiae cells were treated with the antifungal, fluconazole, and expression profiling was conducted under different biological conditions using Affymetrix genome-wide microarrays. Transcripts were filtered with a formal network-based method, sparse simultaneous equation models and Lasso regression (SSEM-Lasso), under different network training conditions. Gene expression results were evaluated using both gene set and single gene target analyses, and the drug's transcriptional effects were narrowed first by pathway and then by individual genes. Variables included: (i) Testing conditions--exposure time and concentration and (ii) Network training conditions--training compendium modifications. Two analyses of SSEM-Lasso output--gene set and single gene--were conducted to gain a better understanding of how SSEM-Lasso predicts perturbation targets.
Conclusions: This study demonstrates that genome-wide microarrays can be optimized using a two-stage strategy for a more in-depth understanding of how a cell manifests biological reactions to a drug treatment at the transcription level. Additionally, a more detailed understanding of how the statistical model, SSEM-Lasso, propagates perturbations through a network of gene regulatory interactions is achieved.
Figures
Similar articles
-
Predicting gene targets of perturbations via network-based filtering of mRNA expression compendia.Bioinformatics. 2008 Nov 1;24(21):2482-90. doi: 10.1093/bioinformatics/btn476. Epub 2008 Sep 8. Bioinformatics. 2008. PMID: 18779235 Free PMC article.
-
Inferring gene targets of drugs and chemical compounds from gene expression profiles.Bioinformatics. 2016 Jul 15;32(14):2120-7. doi: 10.1093/bioinformatics/btw148. Epub 2016 Mar 18. Bioinformatics. 2016. PMID: 27153589 Free PMC article.
-
Genome-wide expression patterns in Saccharomyces cerevisiae: comparison of drug treatments and genetic alterations affecting biosynthesis of ergosterol.Antimicrob Agents Chemother. 2000 May;44(5):1255-65. doi: 10.1128/AAC.44.5.1255-1265.2000. Antimicrob Agents Chemother. 2000. PMID: 10770760 Free PMC article.
-
Saccharomyces cerevisiae and DNA microarray analyses: what did we learn from it for a better understanding and exploitation of yeast biotechnology?Appl Microbiol Biotechnol. 2010 Jun;87(2):391-400. doi: 10.1007/s00253-010-2582-7. Epub 2010 Apr 23. Appl Microbiol Biotechnol. 2010. PMID: 20414652 Review.
-
Chemical-genetic approaches for exploring the mode of action of natural products.Prog Drug Res. 2008;66:237, 239-71. doi: 10.1007/978-3-7643-8595-8_5. Prog Drug Res. 2008. PMID: 18416308 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
