

The functions of cardiolipin in cellular metabolism-potential modifiers of the Barth syndrome phenotype
- PMID: 24445246
- PMCID: PMC4342993
- DOI: 10.1016/j.chemphyslip.2013.12.009
The functions of cardiolipin in cellular metabolism-potential modifiers of the Barth syndrome phenotype
Abstract
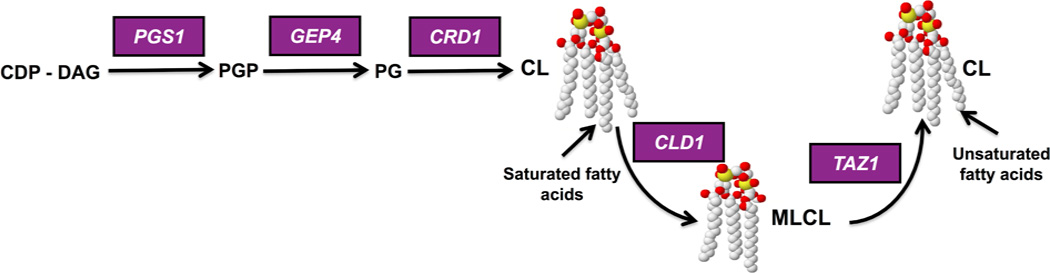
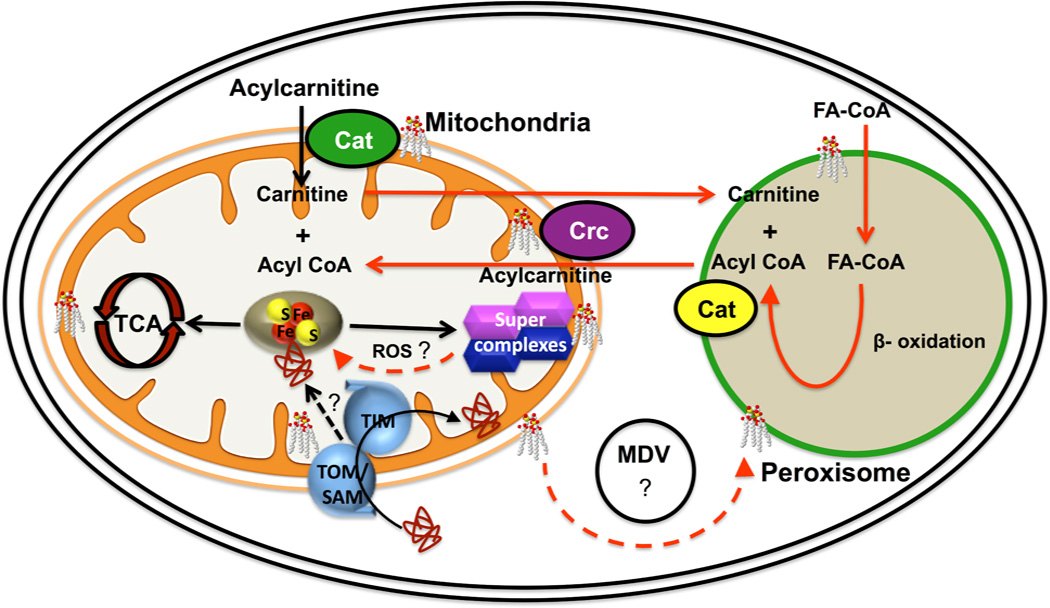
The phospholipid cardiolipin (CL) plays a role in many cellular functions and signaling pathways both inside and outside of mitochondria. This review focuses on the role of CL in energy metabolism. Many reactions of electron transport and oxidative phosphorylation, the transport of metabolites required for these processes, and the stabilization of electron transport chain supercomplexes require CL. Recent studies indicate that CL is required for the synthesis of iron-sulfur (Fe-S) co-factors, which are essential for numerous metabolic pathways. Activation of carnitine shuttle enzymes that are required for fatty acid metabolism is CL dependent. The presence of substantial amounts of CL in the peroxisomal membrane suggests that CL may be required for peroxisomal functions. Understanding the role of CL in energy metabolism may identify physiological modifiers that exacerbate the loss of CL and underlie the variation in symptoms observed in Barth syndrome, a genetic disorder of CL metabolism.
Keywords: Barth syndrome; Bioenergetics; Carnitine transport; Fatty acid utilization; Iron–sulfur biogenesis.
Copyright © 2014 Elsevier Ireland Ltd. All rights reserved.
Figures
References
-
- Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. Journal of bioenergetics and biomembranes. 1999;31:347–366. - PubMed
-
- Bartlett K, Eaton S. Mitochondrial beta-oxidation. European journal of biochemistry / FEBS. 2004;271:462–469. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
