

Predicting DNA methylation level across human tissues
- PMID: 24445802
- PMCID: PMC3973306
- DOI: 10.1093/nar/gkt1380
Predicting DNA methylation level across human tissues
Abstract
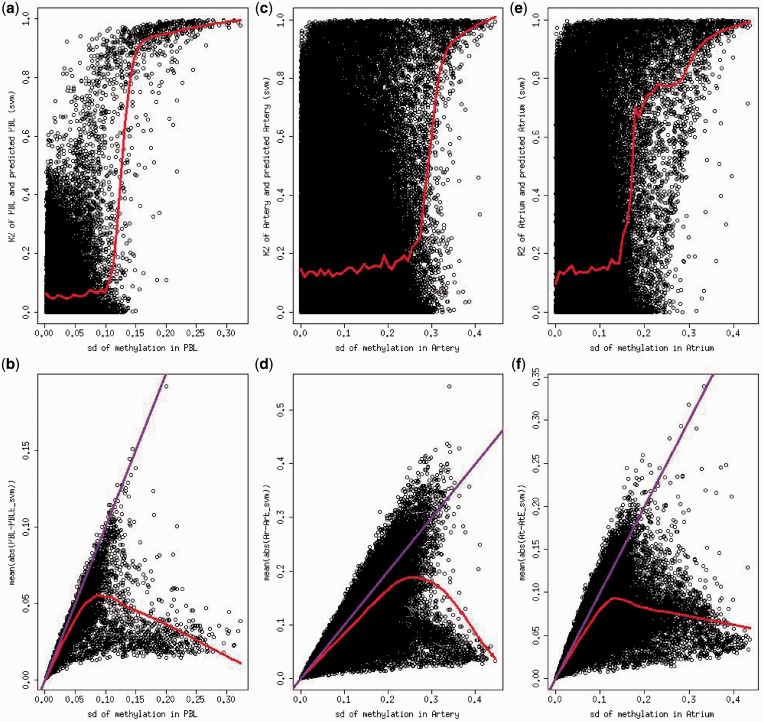
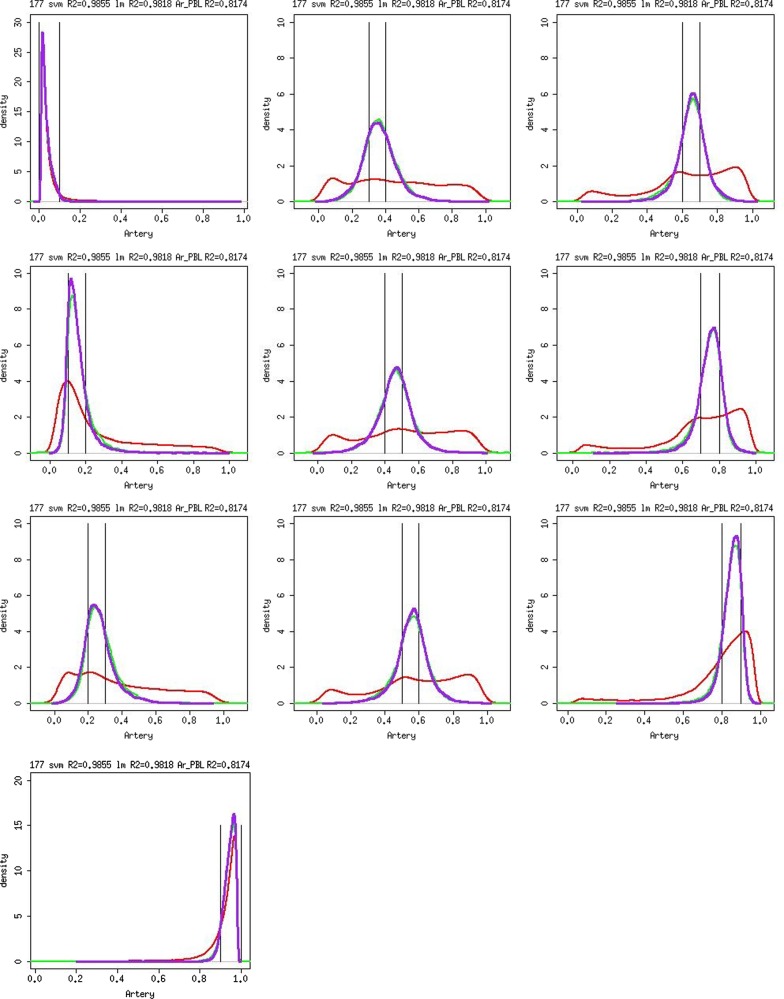
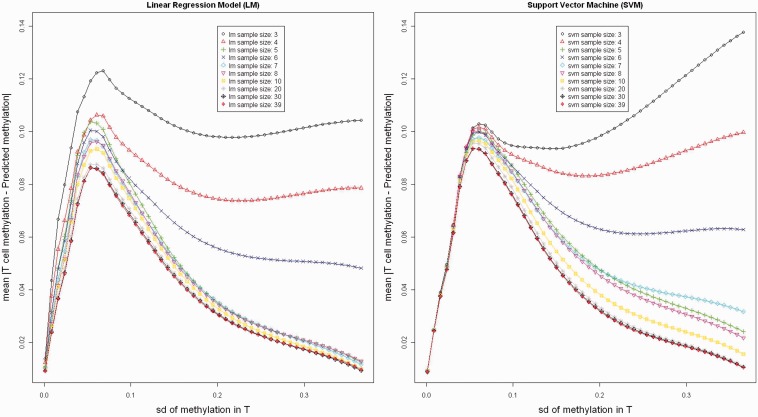
Differences in methylation across tissues are critical to cell differentiation and are key to understanding the role of epigenetics in complex diseases. In this investigation, we found that locus-specific methylation differences between tissues are highly consistent across individuals. We developed a novel statistical model to predict locus-specific methylation in target tissue based on methylation in surrogate tissue. The method was evaluated in publicly available data and in two studies using the latest IlluminaBeadChips: a childhood asthma study with methylation measured in both peripheral blood leukocytes (PBL) and lymphoblastoid cell lines; and a study of postoperative atrial fibrillation with methylation in PBL, atrium and artery. We found that our method can greatly improve accuracy of cross-tissue prediction at CpG sites that are variable in the target tissue [R(2) increases from 0.38 (original R(2) between tissues) to 0.89 for PBL-to-artery prediction; from 0.39 to 0.95 for PBL-to-atrium; and from 0.81 to 0.98 for lymphoblastoid cell line-to-PBL based on cross-validation, and confirmed using cross-study prediction]. An extended model with multiple CpGs further improved performance. Our results suggest that large-scale epidemiology studies using easy-to-access surrogate tissues (e.g. blood) could be recalibrated to improve understanding of epigenetics in hard-to-access tissues (e.g. atrium) and might enable non-invasive disease screening using epigenetic profiles.
Figures



References
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
