

Adaptor protein2 (AP2) orchestrates CXCR2-mediated cell migration
- PMID: 24450359
- PMCID: PMC3966550
- DOI: 10.1111/tra.12154
Adaptor protein2 (AP2) orchestrates CXCR2-mediated cell migration
Abstract
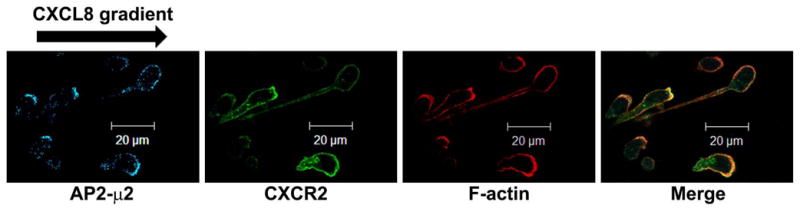
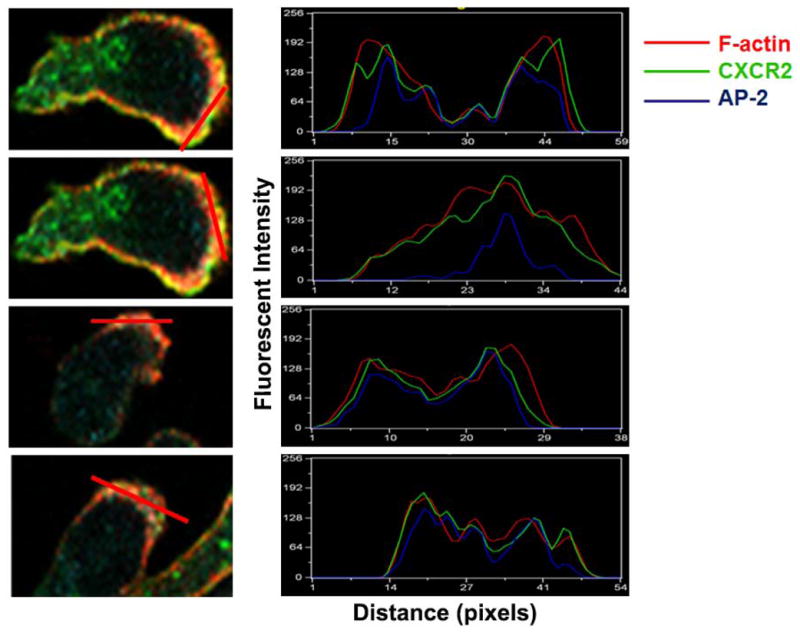
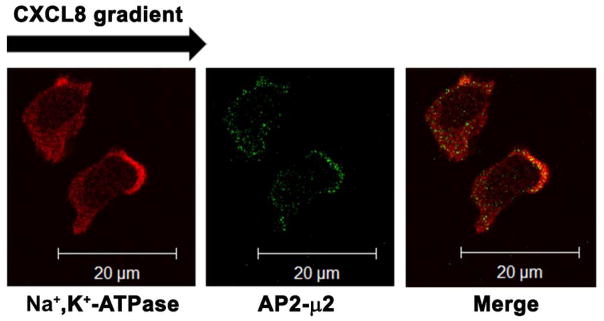
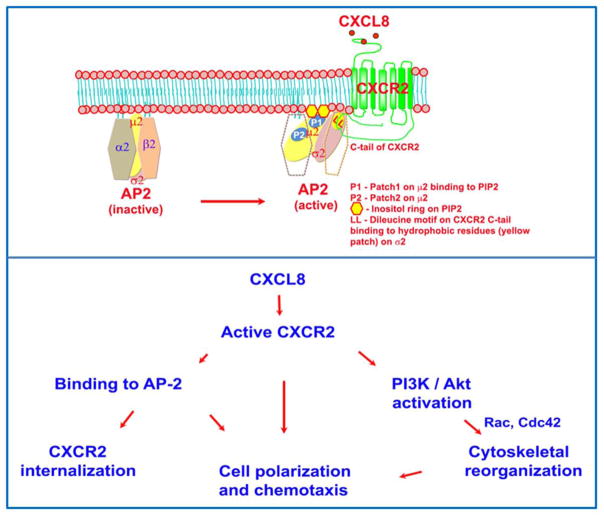
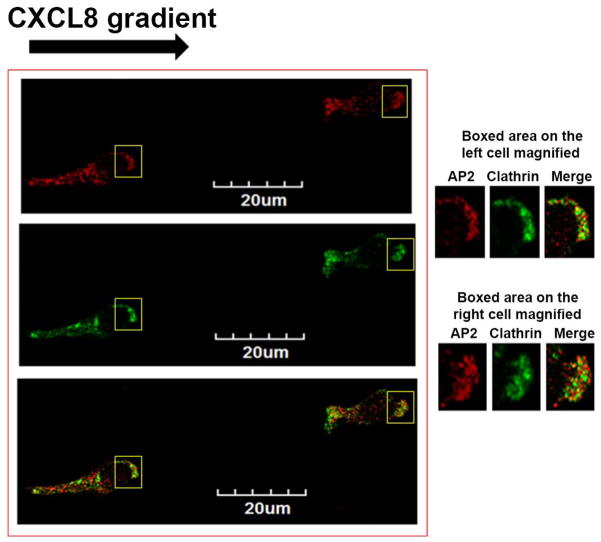
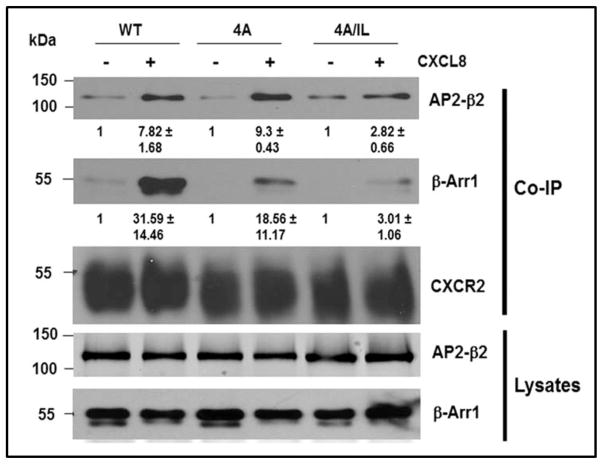
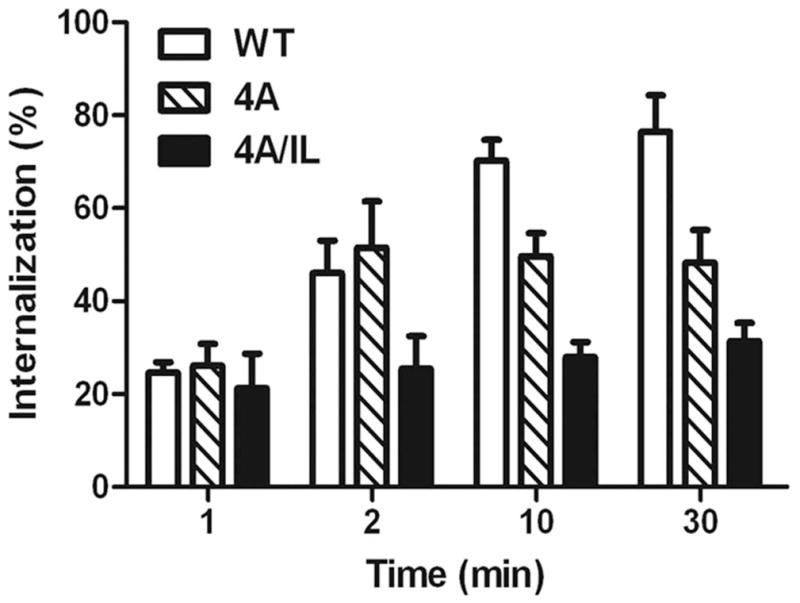
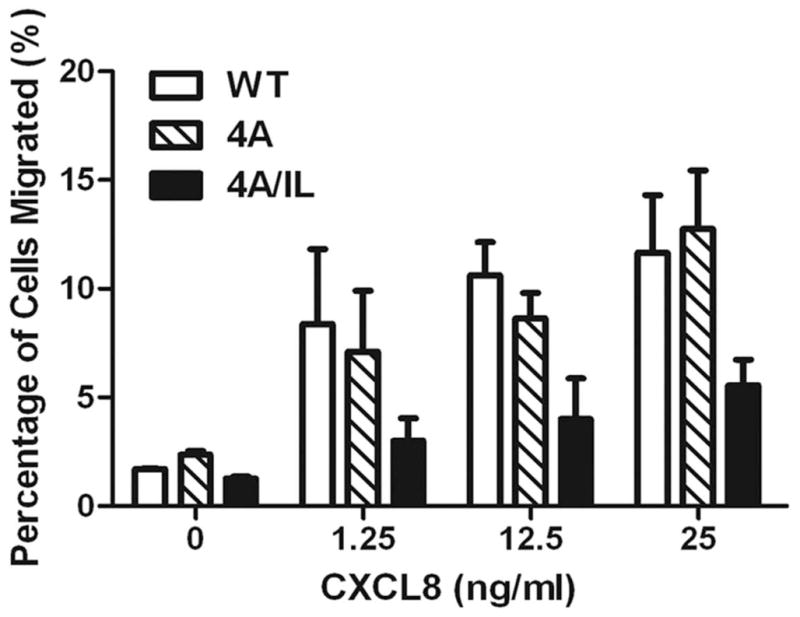
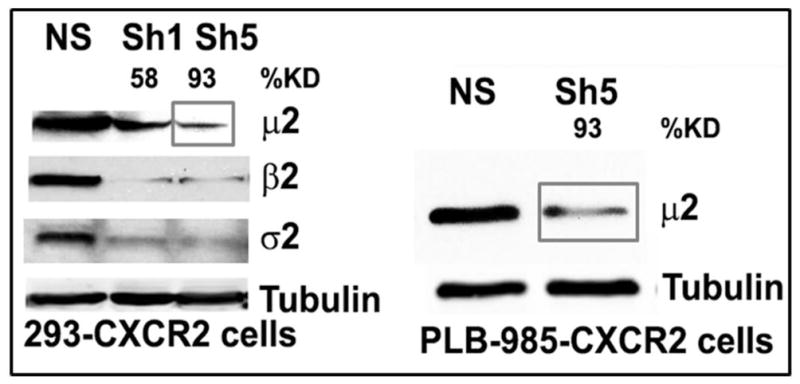
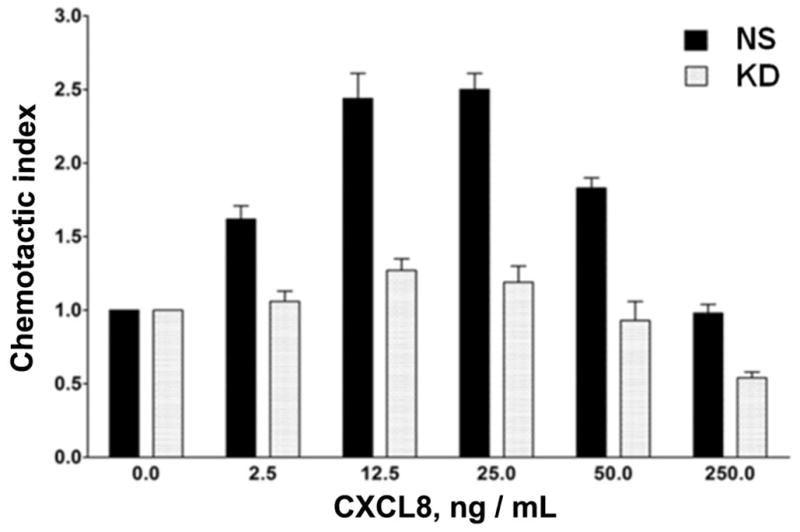
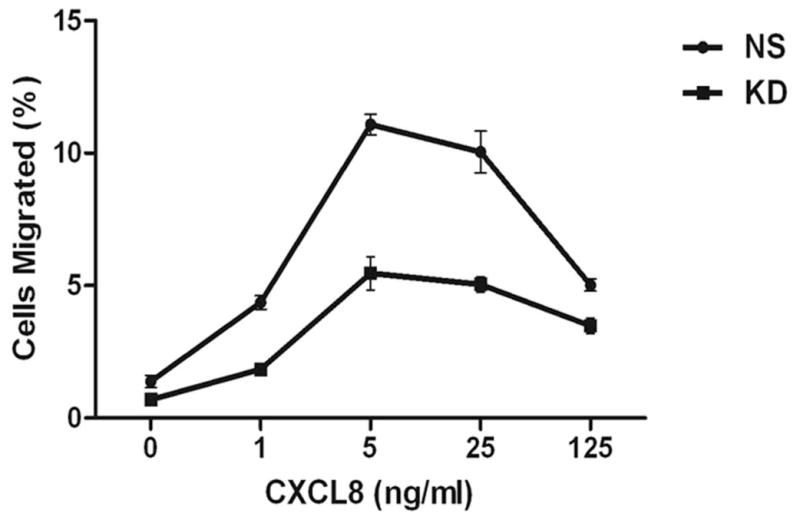
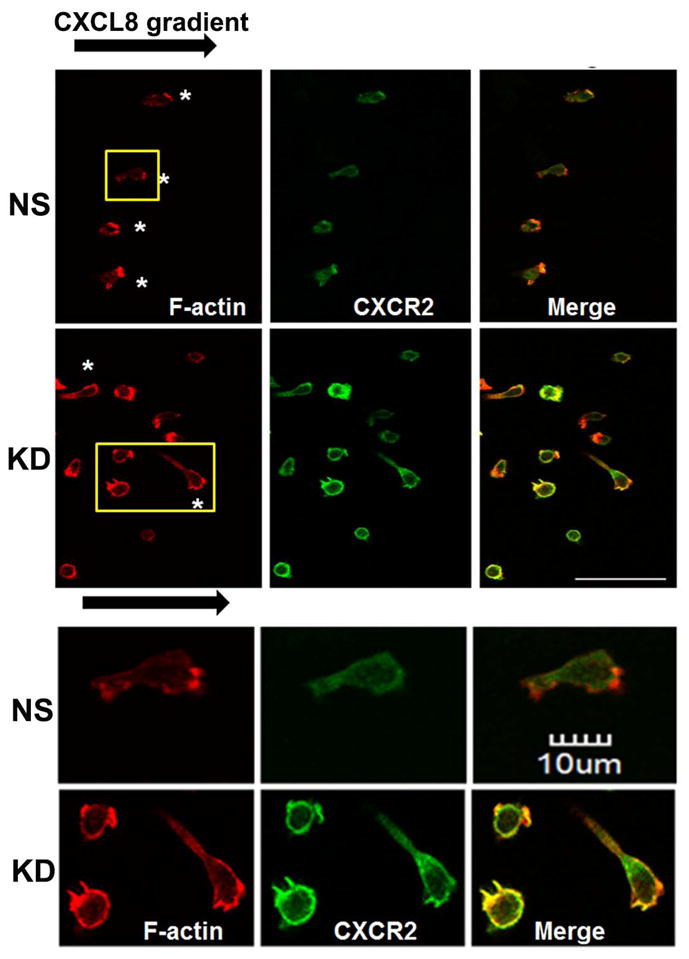
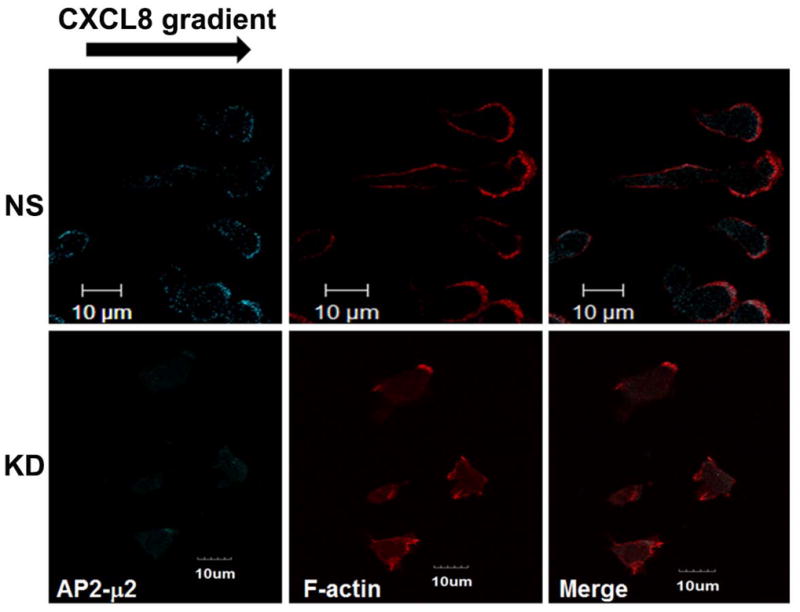
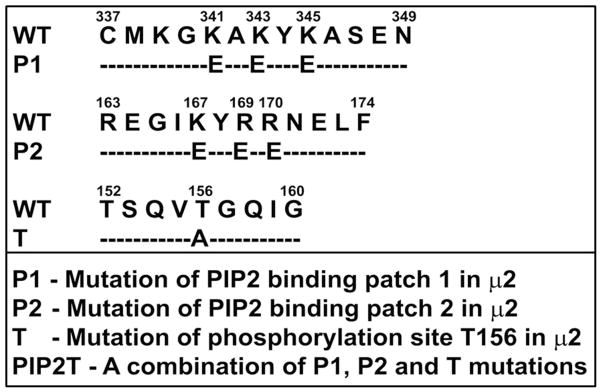
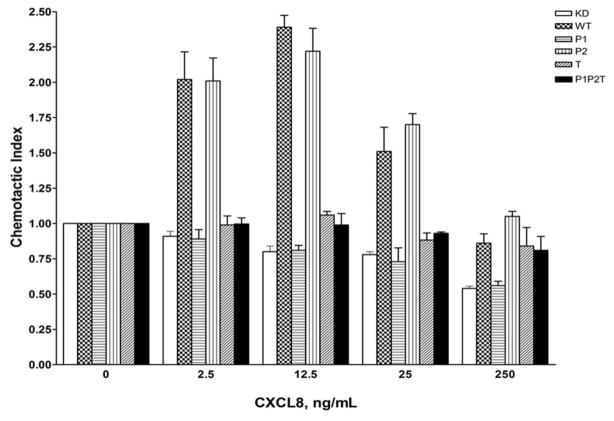
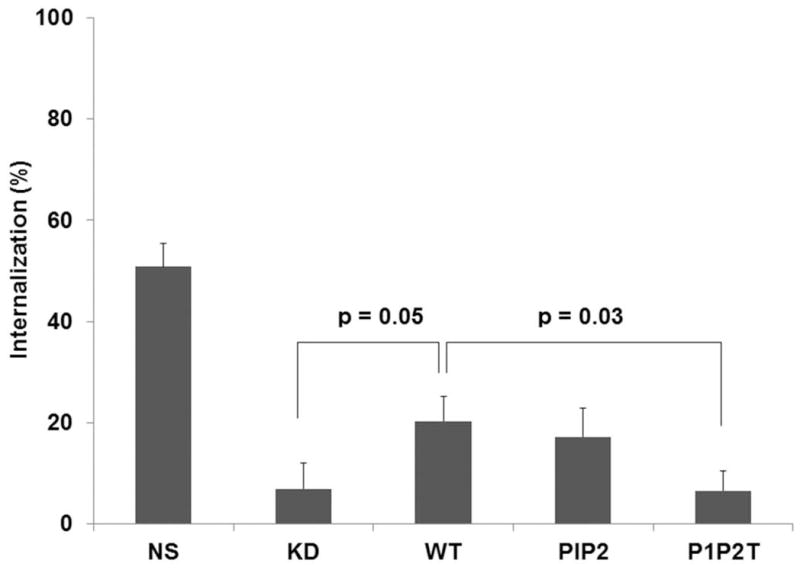
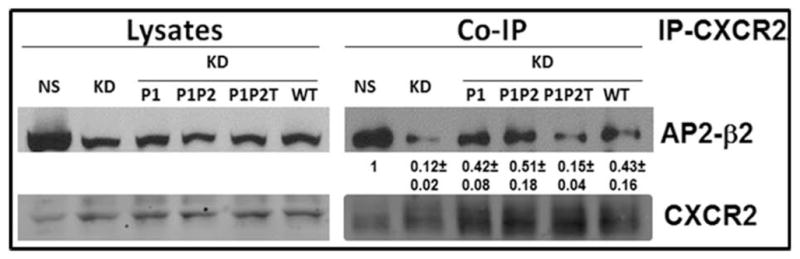
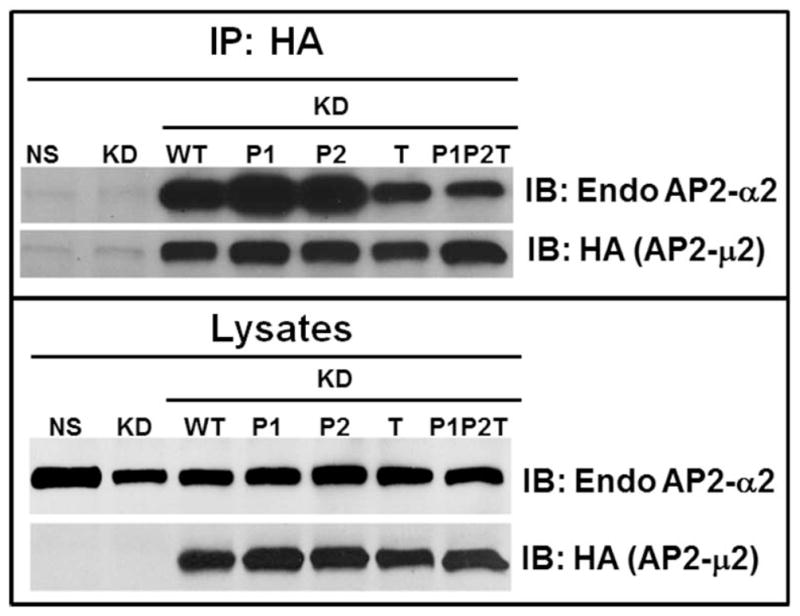
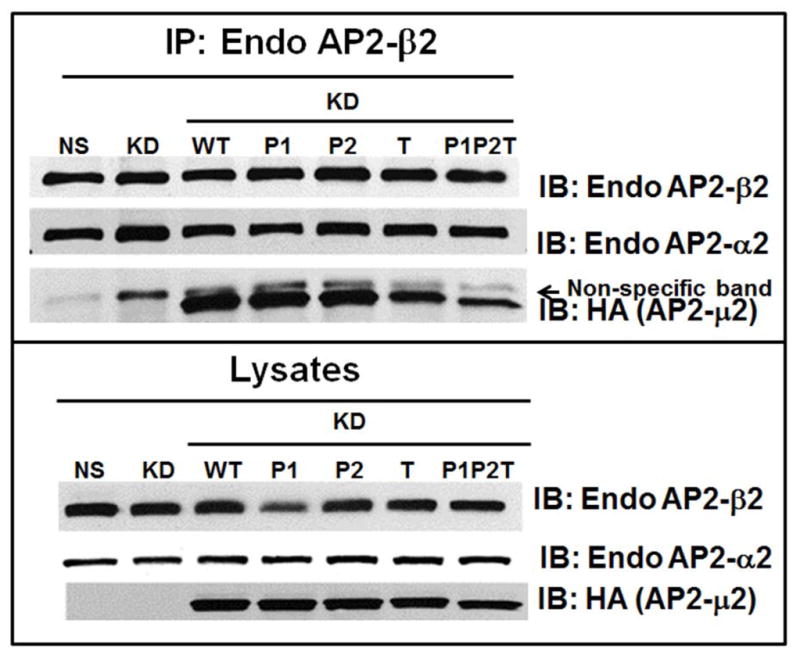
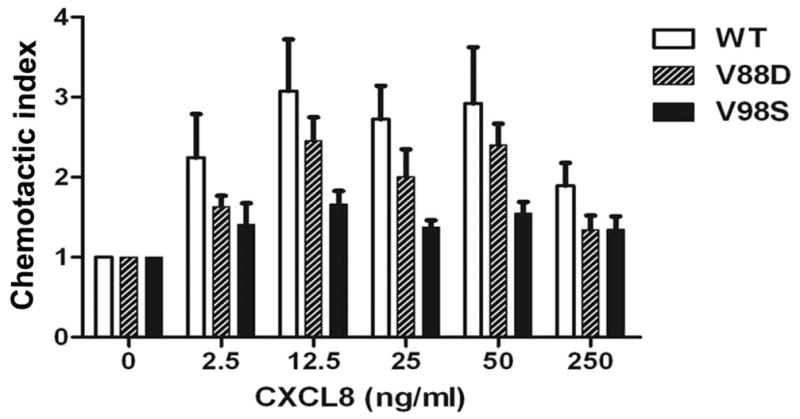
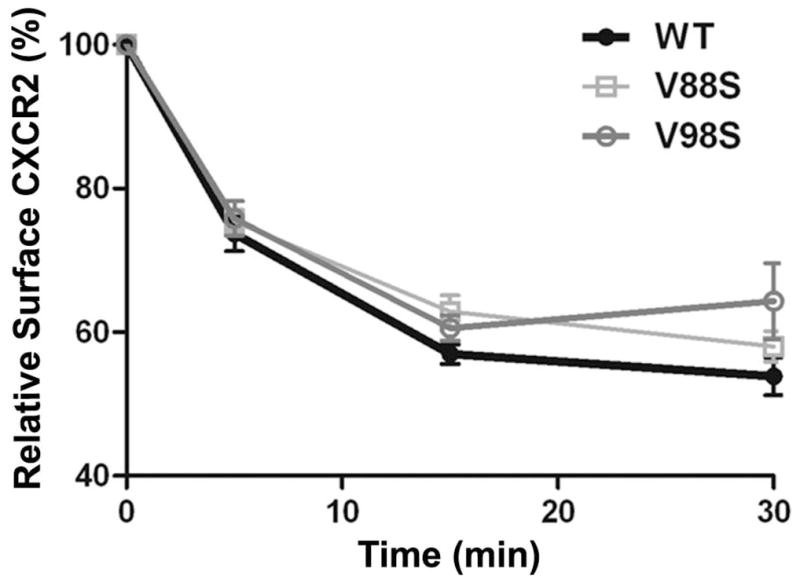
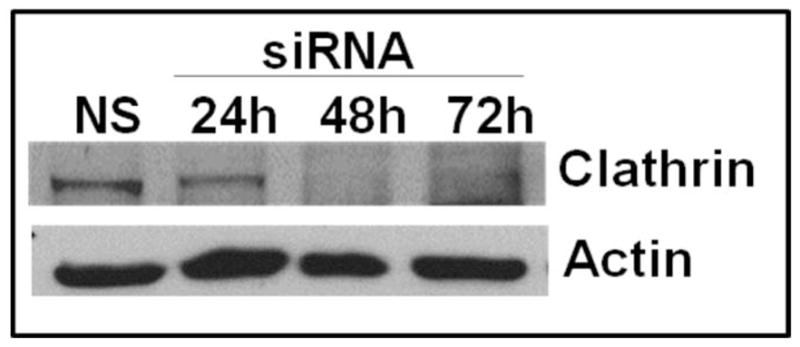
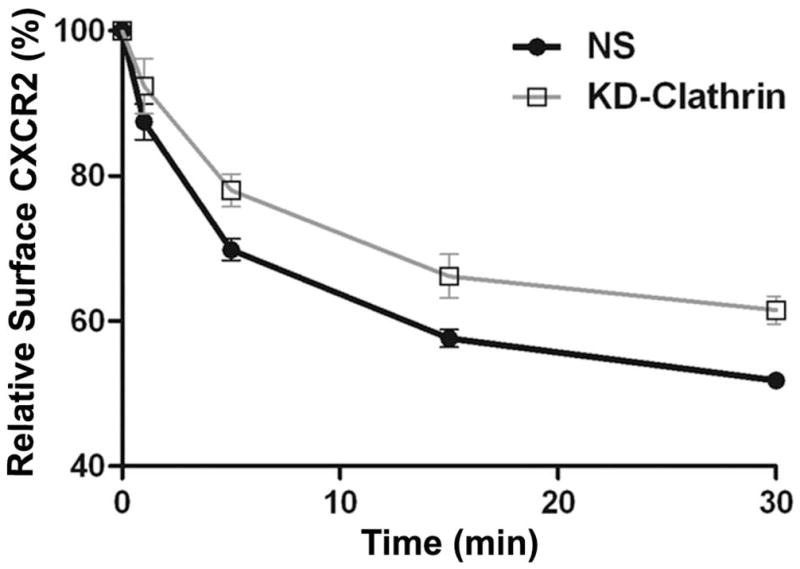
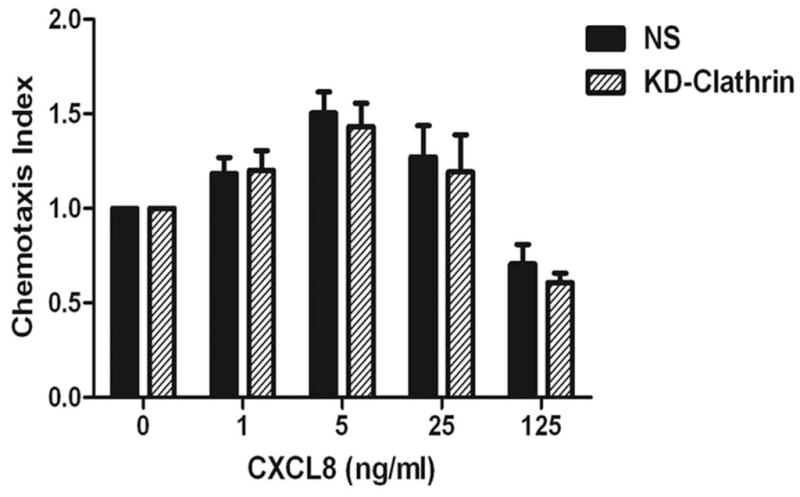
The chemokine receptor CXCR2 is vital for inflammation, wound healing, angiogenesis, cancer progression and metastasis. Adaptor protein 2 (AP2), a clathrin binding heterotetrameric protein comprised of α, β2, μ2 and σ2 subunits, facilitates clathrin-mediated endocytosis. Mutation of the LLKIL motif in the CXCR2 carboxyl-terminal domain (CTD) results in loss of AP2 binding to the receptor and loss of ligand-mediated receptor internalization and chemotaxis. AP2 knockdown also results in diminished ligand-mediated CXCR2 internalization, polarization and chemotaxis. Using knockdown/rescue approaches with AP2-μ2 mutants, the binding domains were characterized in reference to CXCR2 internalization and chemotaxis. When in an open conformation, μ2 Patch 1 and Patch 2 domains bind tightly to membrane PIP2 phospholipids. When AP2-μ2, is replaced with μ2 mutated in Patch 1 and/or Patch 2 domains, ligand-mediated receptor binding and internalization are not lost. However, chemotaxis requires AP2-μ2 Patch 1, but not Patch 2. AP2-σ2 has been demonstrated to bind dileucine motifs to facilitate internalization. Expression of AP2-σ2 V88D and V98S dominant negative mutants resulted in loss of CXCR2 mediated chemotaxis. Thus, AP2 binding to both membrane phosphatidylinositol phospholipids and dileucine motifs is crucial for directional migration or chemotaxis. Moreover, AP2-mediated receptor internalization can be dissociated from AP2-mediated chemotaxis.
Keywords: AP2; AP2-μ2; AP2-σ2; CXCR2; PIP2 patches; chemotaxis; internalization.
© 2014 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Figures

References
-
- Arai H, Charo IF. Differential regulation of G-protein-mediated signaling by chemokine receptors. The Journal of biological chemistry. 1996;271(36):21814–21819. - PubMed
-
- Addison CL, Daniel TO, Burdick MD, Liu H, Ehlert JE, Xue YY, Buechi L, Walz A, Richmond A, Strieter RM. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol. 2000;165(9):5269–5277. - PubMed
-
- Matsuo Y, Raimondo M, Woodward TA, Wallace MB, Gill KR, Tong Z, Burdick MD, Yang Z, Strieter RM, Hoffman RM, Guha S. CXC-chemokine/CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer. Int J Cancer. 2009;125(5):1027–1037. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
