

Genome resolved analysis of a premature infant gut microbial community reveals a Varibaculum cambriense genome and a shift towards fermentation-based metabolism during the third week of life
- PMID: 24451181
- PMCID: PMC4177395
- DOI: 10.1186/2049-2618-1-30
Genome resolved analysis of a premature infant gut microbial community reveals a Varibaculum cambriense genome and a shift towards fermentation-based metabolism during the third week of life
Abstract
Background: The premature infant gut has low individual but high inter-individual microbial diversity compared with adults. Based on prior 16S rRNA gene surveys, many species from this environment are expected to be similar to those previously detected in the human microbiota. However, the level of genomic novelty and metabolic variation of strains found in the infant gut remains relatively unexplored.
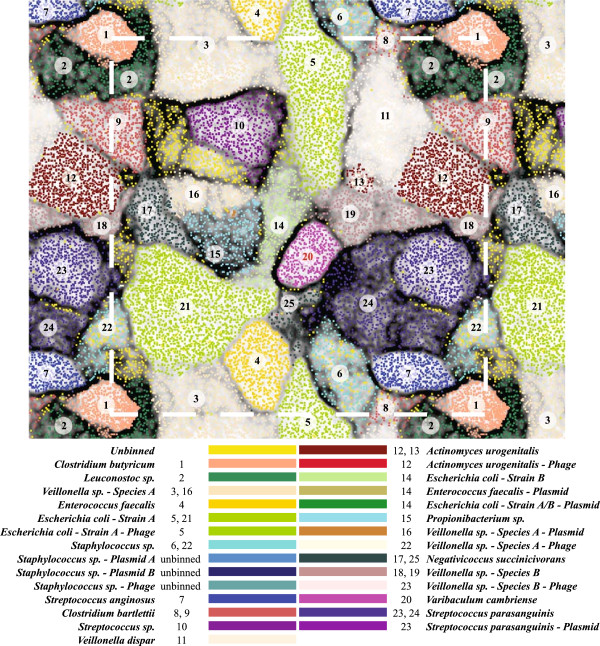
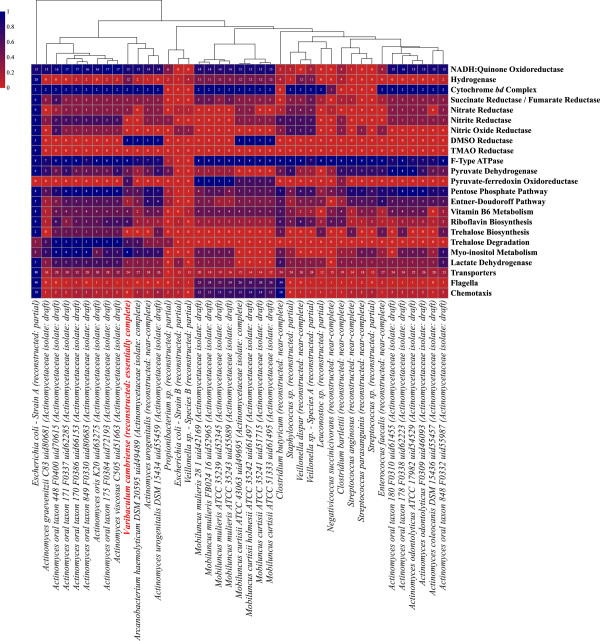
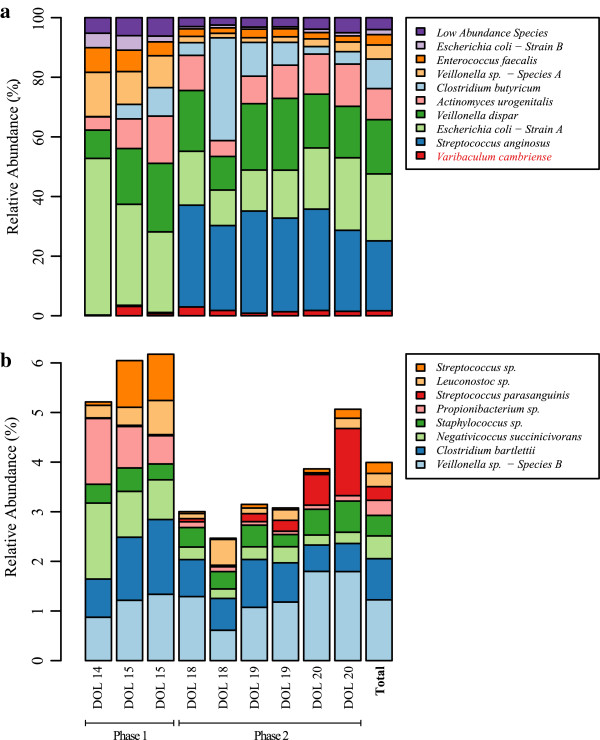
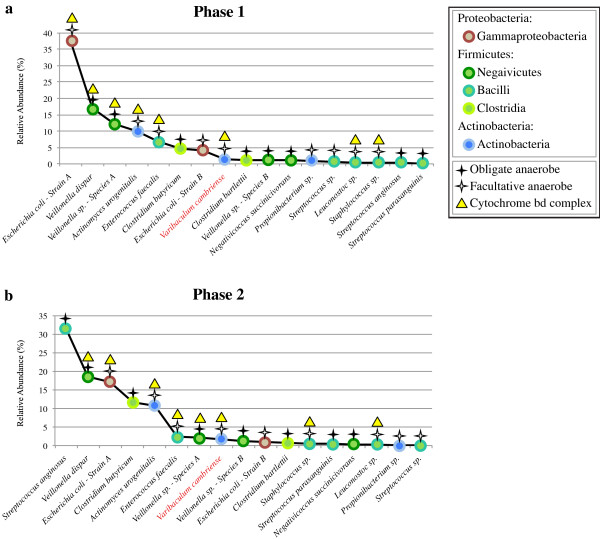
Results: To study the stability and function of early microbial colonizers of the premature infant gut, nine stool samples were taken during the third week of life of a premature male infant delivered via Caesarean section. Metagenomic sequences were assembled and binned into near-complete and partial genomes, enabling strain-level genomic analysis of the microbial community.We reconstructed eleven near-complete and six partial bacterial genomes representative of the key members of the microbial community. Twelve of these genomes share >90% putative ortholog amino acid identity with reference genomes. Manual curation of the assembly of one particularly novel genome resulted in the first essentially complete genome sequence (in three pieces, the order of which could not be determined due to a repeat) for Varibaculum cambriense (strain Dora), a medically relevant species that has been implicated in abscess formation.During the period studied, the microbial community undergoes a compositional shift, in which obligate anaerobes (fermenters) overtake Escherichia coli as the most abundant species. Other species remain stable, probably due to their ability to either respire anaerobically or grow by fermentation, and their capacity to tolerate fluctuating levels of oxygen. Metabolic predictions for V. cambriense suggest that, like other members of the microbial community, this organism is able to process various sugar substrates and make use of multiple different electron acceptors during anaerobic respiration. Genome comparisons within the family Actinomycetaceae reveal important differences related to respiratory metabolism and motility.
Conclusions: Genome-based analysis provided direct insight into strain-specific potential for anaerobic respiration and yielded the first genome for the genus Varibaculum. Importantly, comparison of these de novo assembled genomes with closely related isolate genomes supported the accuracy of the metagenomic methodology. Over a one-week period, the early gut microbial community transitioned to a community with a higher representation of obligate anaerobes, emphasizing both taxonomic and metabolic instability during colonization.
Figures
References
-
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J-M, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P. et al.A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. - DOI - PMC - PubMed
-
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
