

Review
doi: 10.1038/nrm3741.
Regulated protein turnover: snapshots of the proteasome in action
Affiliations
- PMID: 24452470
- PMCID: PMC4384331
- DOI: 10.1038/nrm3741
Item in Clipboard
Review
Regulated protein turnover: snapshots of the proteasome in action
Nat Rev Mol Cell Biol.
2014 Feb.
Abstract
The ubiquitin proteasome system (UPS) is the main ATP-dependent protein degradation pathway in the cytosol and nucleus of eukaryotic cells. At its centre is the 26S proteasome, which degrades regulatory proteins and misfolded or damaged proteins. In a major breakthrough, several groups have determined high-resolution structures of the entire 26S proteasome particle in different nucleotide conditions and with and without substrate using cryo-electron microscopy combined with other techniques. These structures provide some surprising insights into the functional mechanism of the proteasome and will give invaluable guidance for genetic and biochemical studies of this key regulatory system.
Figures
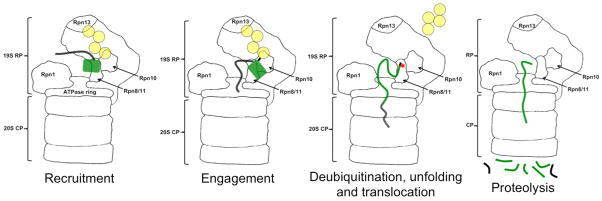
The proteasome recognizes ubiquitin tags in substrates through its receptors, here Rpn10 and Rpn13, and then initiates degradation at an unstructured region in the substrate. As the ATPase motors pull the substrate into the degradation channel, the ubiquitin chain is cleaved off, the substrate unfolds and is finally cleaved into peptides.
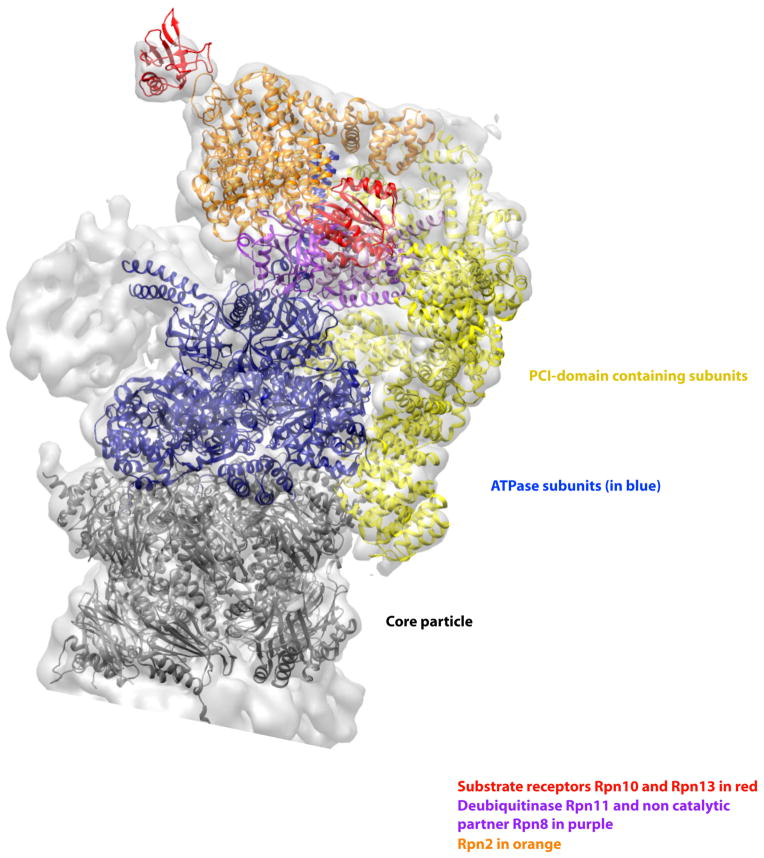
The structure of the proteasome in the presence of ATP but without substrate shows the six PCI domain containing subunits (shown in yellow) form a U-shaped structure that interacts with many subunits in the RP and a ring of α-subunits in the core particle (shown in dark grey). These interactions probably stabilize the RP and its interaction with the proteolytic core. They may also provide a mechanism for cooperative interaction of the different biochemical activities of the various subunits. The six ATPase subunits are shown in blue, the ubiquitin receptors Rpn10 and Rpn13 are shown in red, the organizing subunit Rpn2 in orange, and the metallo protease subunits Rpn8/11 heterodimer is in light purple. The figure was generated from the EM structure (EMD-1992) from Lander et al., 2012 and atomic coordinates from Beck et al., 2012 pdb file (PDB code: 4B4T), . All models were generated and visualized with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). The crystal structures (pdb ID 4B4T) were fitted into the experimental electron microscopy map (EMDB 1992) with the algorithms provided by Chimera and corrected manually. The Segger package was used for the segmentation of the electron density map.
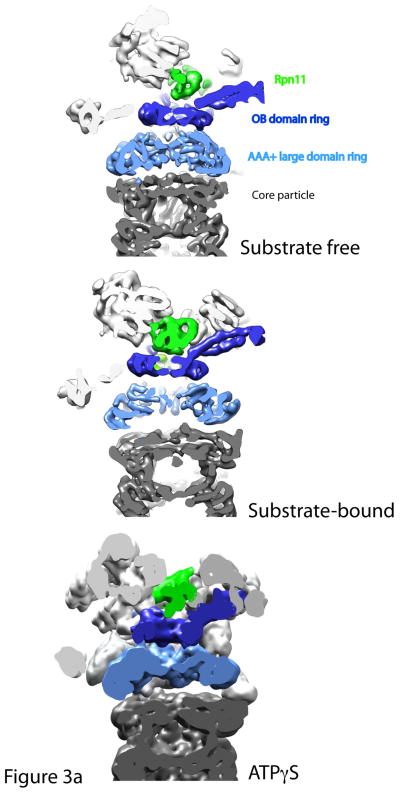
Cutaway views of the central channels of the proteasome in the substrate-free, substrate-bound and ATPγS conditions. 3a: A continuous translocation channel forms upon ATPγS or substrate binding : Top is the substrate-free condition, where the OB domain ring (dark blue), the AAA+ domain ring (light blue) and the entrance to the core particle (grey) are all slightly out of alignment. Rpn11 (green) sits to the side of the OB domain ring. However, upon substrate binding or ATPγS conditions, the rings shift and tilt such that the central pore of each of the rings is aligned coaxially. Thus, a continuous central channel forms in the bottom two structures. Rpn11 moves directly over the OB-ring in both the substrate-bound and ATPγS conditions, aligning its catalytic active site with the translocation channel and possibly preventing substrate escape. 3b: Central pore of AAA+ domain ring expands considerably after substrate binding : Cutaway views looking directly into the translocation channel from the top of the proteasome show that the central pore of the AAA+ ring expands upon substrate binding to allow for substrate entry. AAA+ domain ring is shown in light blue. The figure was generated from the EM maps from Lander et al., 2012 (EMD-1992), Matyskiela et al 2013 (EMD-5669) and Sledz et al 2013 (EMD-2348), , . All models were generated and visualized with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). The EM map from Sledz et al 2013 (EMD-2348) was segmented using the Segger package, .
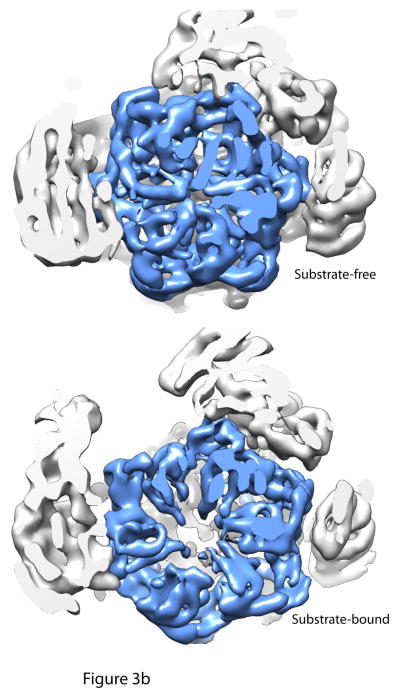
Cutaway views of the central channels of the proteasome in the substrate-free, substrate-bound and ATPγS conditions. 3a: A continuous translocation channel forms upon ATPγS or substrate binding : Top is the substrate-free condition, where the OB domain ring (dark blue), the AAA+ domain ring (light blue) and the entrance to the core particle (grey) are all slightly out of alignment. Rpn11 (green) sits to the side of the OB domain ring. However, upon substrate binding or ATPγS conditions, the rings shift and tilt such that the central pore of each of the rings is aligned coaxially. Thus, a continuous central channel forms in the bottom two structures. Rpn11 moves directly over the OB-ring in both the substrate-bound and ATPγS conditions, aligning its catalytic active site with the translocation channel and possibly preventing substrate escape. 3b: Central pore of AAA+ domain ring expands considerably after substrate binding : Cutaway views looking directly into the translocation channel from the top of the proteasome show that the central pore of the AAA+ ring expands upon substrate binding to allow for substrate entry. AAA+ domain ring is shown in light blue. The figure was generated from the EM maps from Lander et al., 2012 (EMD-1992), Matyskiela et al 2013 (EMD-5669) and Sledz et al 2013 (EMD-2348), , . All models were generated and visualized with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). The EM map from Sledz et al 2013 (EMD-2348) was segmented using the Segger package, .
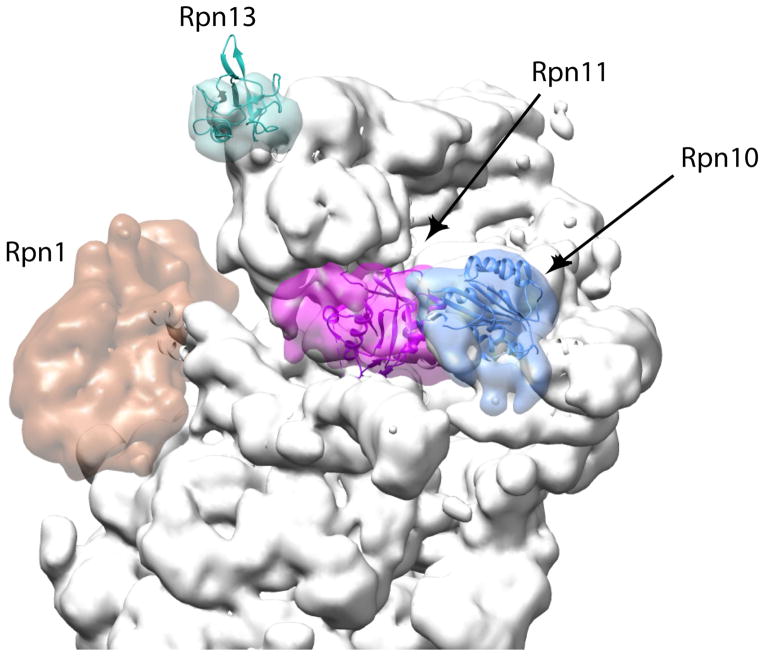
The locations of the two well-characterized ubiquitin receptors on the proteasome, Rpn10 (blue) and Rpn13 (cyan), together with the organizing subunit Rpn1 (dark orange) and the deubiquitinase Rpn11 (purple) are highlighted. The remaining proteasome subunits are shown in grey. The locations of Rpn10 and Rpn13 might allow a polyubiquitin chain to bind both receptors simultaneously or they serve as an adaptive platform to feed structurally diverse substrates into the translocation motor (left). Rpn1 provides a docking site for UbL-UBA proteins such as Rad23 that deliver substrates to the proteasome. The figure was generated using the EM structure (EMD-1992) from Lander et al 2012 and atomic coordinates from Beck 2012 pdb file (PDB code: 4B4T), using Chimera and Segger as in Figure 2, .
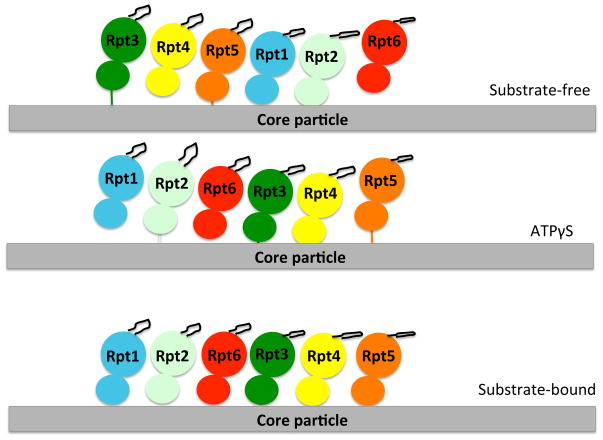
A cartoon representation of the arrangement of the six ATPases of the proteasome and the aromatic-hydrophobic-glycine loop (black loop from each ATPase structure) for each of the conditions in which proteasome structures were determined. Each ATPase is represented with its large and small domain and in a different color. The spiraled arrangement of the AAA+ domains is obtained in both the substrate-free and ATPγS conditions, though the subunits rearrange in the ATPγS condition, so that now Rpt5 “bridges” the spiral. In the substrate-bound condition, the ATPase domains rearrange to a level plane, but the aromatic-hydrophobic-glycine loops are still arranged in a spiral due to the different tiltings of the ATPase domains. The figure is adapted from Beckwith et al. 2013.
References
-
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. - PubMed
-
- Varshavsky A. The ubiquitin system. Trends Biochem Sci. 1997;22:383–7. - PubMed
-
- Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–47. - PubMed
-
- Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–39. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
