

Coarse-grained molecular simulation of epidermal growth factor receptor protein tyrosine kinase multi-site self-phosphorylation
- PMID: 24453959
- PMCID: PMC3894164
- DOI: 10.1371/journal.pcbi.1003435
Coarse-grained molecular simulation of epidermal growth factor receptor protein tyrosine kinase multi-site self-phosphorylation
Abstract
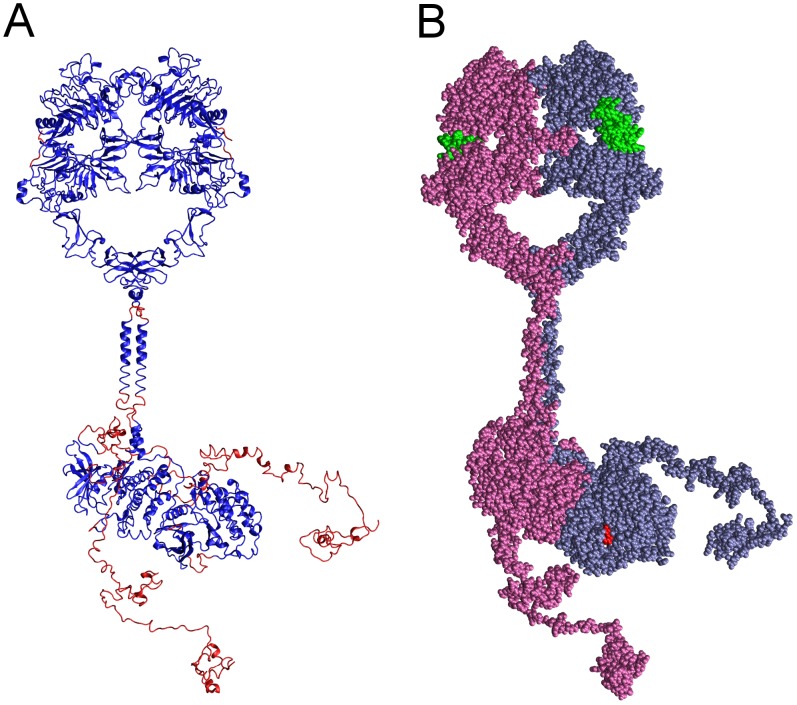
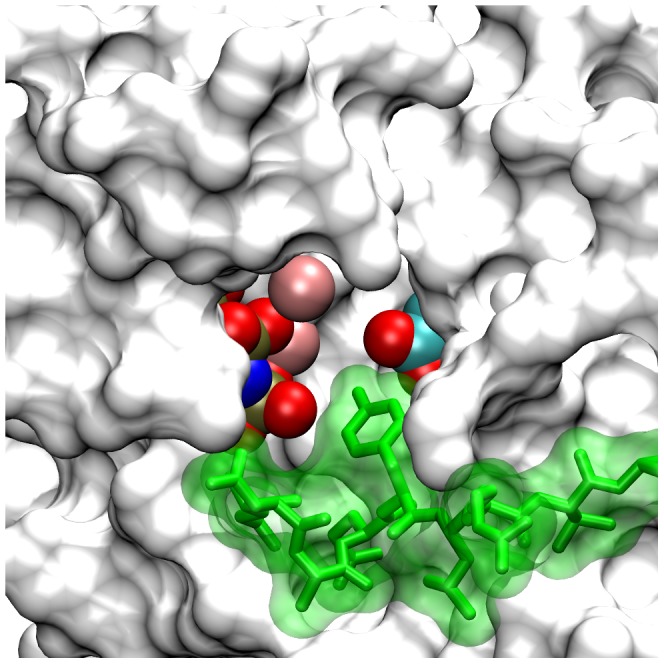
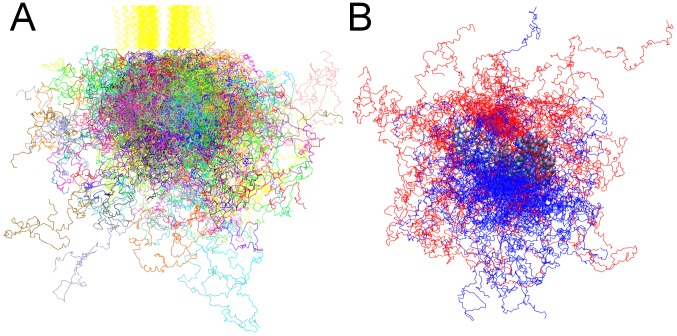
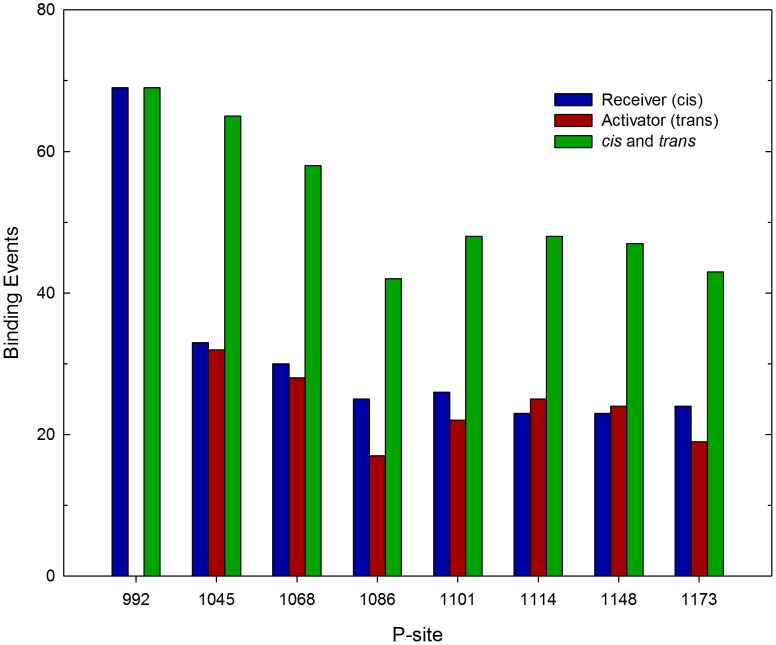
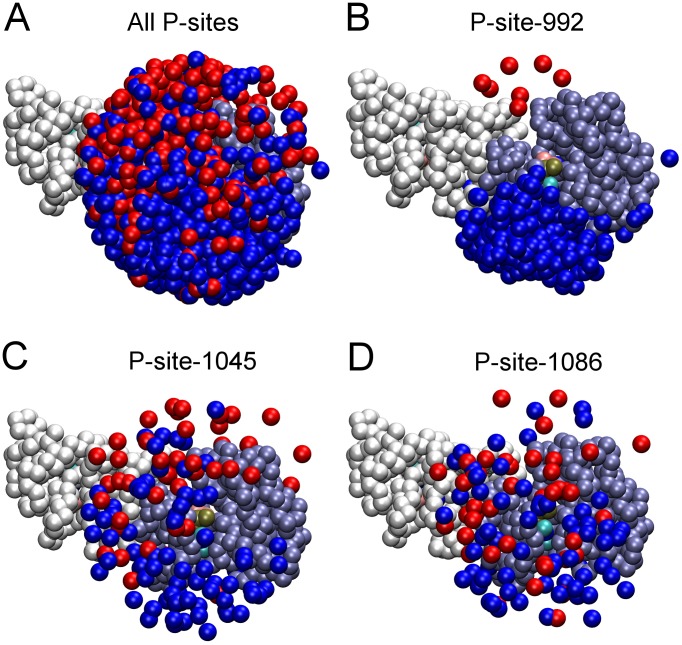
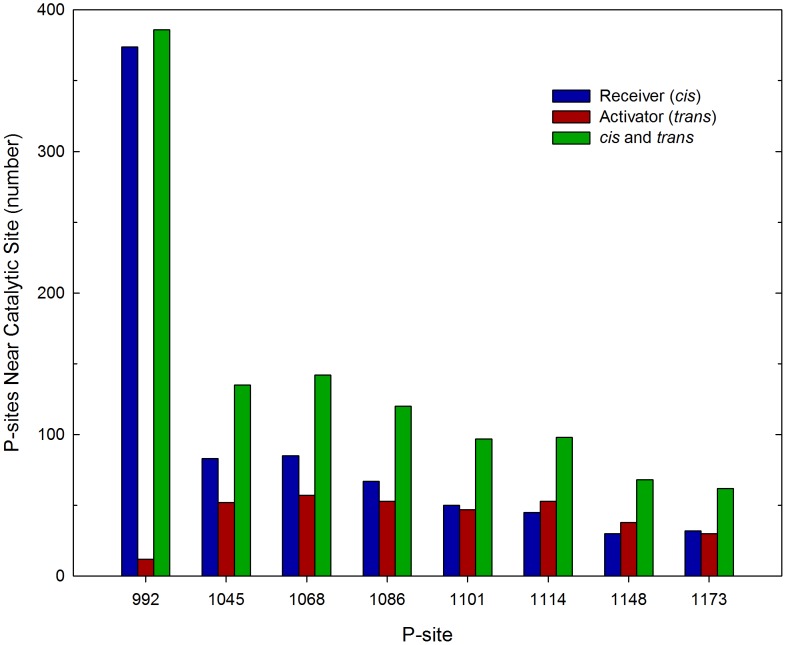
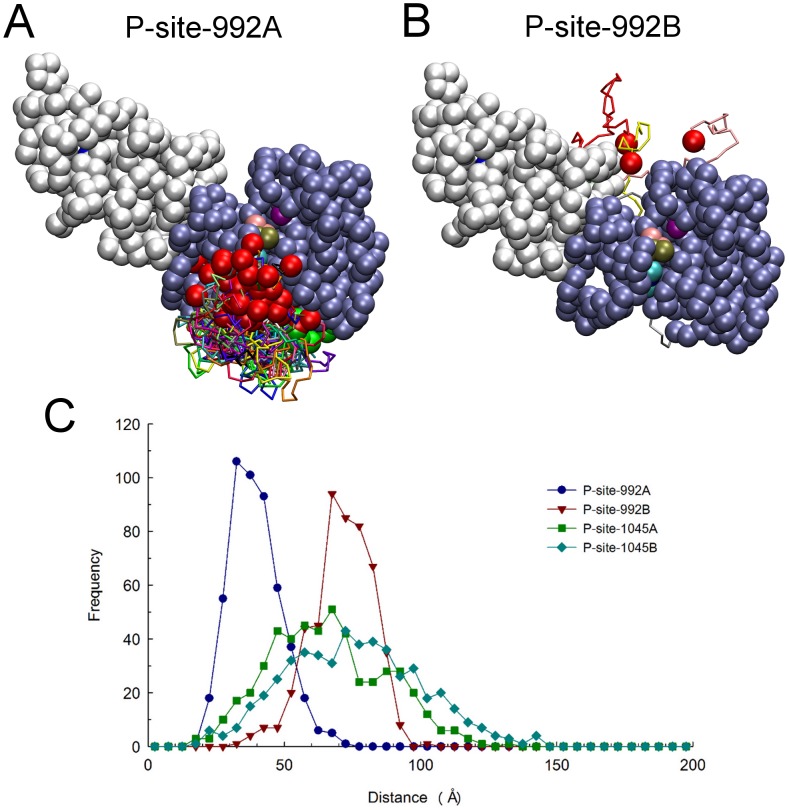
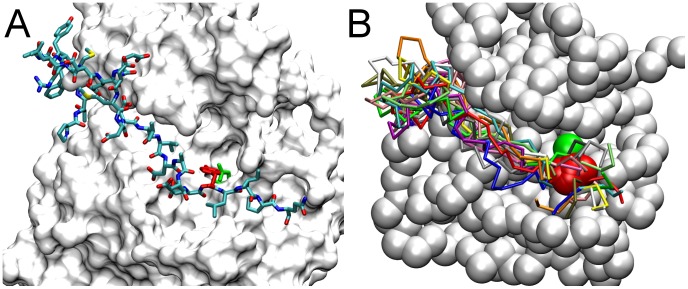
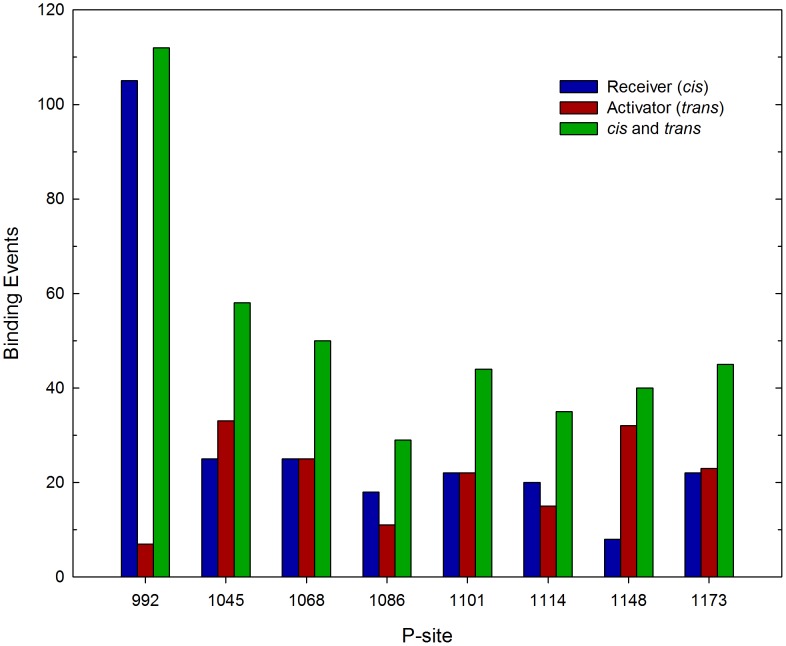
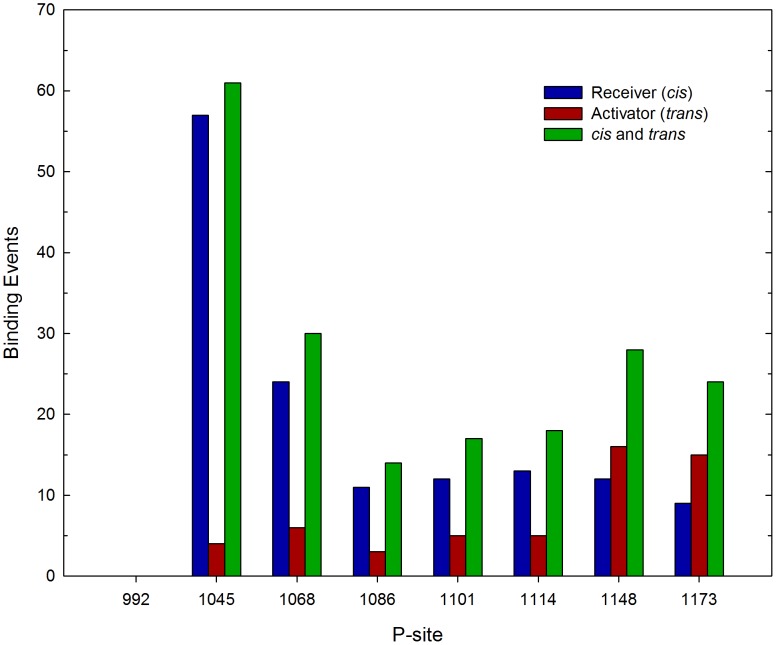
Upon the ligand-dependent dimerization of the epidermal growth factor receptor (EGFR), the intrinsic protein tyrosine kinase (PTK) activity of one receptor monomer is activated, and the dimeric receptor undergoes self-phosphorylation at any of eight candidate phosphorylation sites (P-sites) in either of the two C-terminal (CT) domains. While the structures of the extracellular ligand binding and intracellular PTK domains are known, that of the ∼225-amino acid CT domain is not, presumably because it is disordered. Receptor phosphorylation on CT domain P-sites is critical in signaling because of the binding of specific signaling effector molecules to individual phosphorylated P-sites. To investigate how the combination of conventional substrate recognition and the unique topological factors involved in the CT domain self-phosphorylation reaction lead to selectivity in P-site phosphorylation, we performed coarse-grained molecular simulations of the P-site/catalytic site binding reactions that precede EGFR self-phosphorylation events. Our results indicate that self-phosphorylation of the dimeric EGFR, although generally believed to occur in trans, may well occur with a similar efficiency in cis, with the P-sites of both receptor monomers being phosphorylated to a similar extent. An exception was the case of the most kinase-proximal P-site-992, the catalytic site binding of which occurred exclusively in cis via an intramolecular reaction. We discovered that the in cis interaction of P-site-992 with the catalytic site was facilitated by a cleft between the N-terminal and C-terminal lobes of the PTK domain that allows the short CT domain sequence tethering P-site-992 to the PTK core to reach the catalytic site. Our work provides several new mechanistic insights into the EGFR self-phosphorylation reaction, and demonstrates the potential of coarse-grained molecular simulation approaches for investigating the complexities of self-phosphorylation in molecules such as EGFR (HER/ErbB) family receptors and growth factor receptor PTKs in general.
Conflict of interest statement
The author has declared that no competing interests exist.
Figures
Similar articles
-
A specific amino acid context in EGFR and HER2 phosphorylation sites enables selective binding to the active site of Src homology phosphatase 2 (SHP2).J Biol Chem. 2020 Mar 13;295(11):3563-3575. doi: 10.1074/jbc.RA119.011422. Epub 2020 Feb 4. J Biol Chem. 2020. PMID: 32024694 Free PMC article.
-
Ligand regulates epidermal growth factor receptor kinase specificity: activation increases preference for GAB1 and SHC versus autophosphorylation sites.J Biol Chem. 2004 Sep 10;279(37):38143-50. doi: 10.1074/jbc.M405760200. Epub 2004 Jul 1. J Biol Chem. 2004. PMID: 15231819
-
Co-conserved features associated with cis regulation of ErbB tyrosine kinases.PLoS One. 2010 Dec 13;5(12):e14310. doi: 10.1371/journal.pone.0014310. PLoS One. 2010. PMID: 21179209 Free PMC article.
-
Heterodimerization and functional interaction between EGF receptor family members: a new signaling paradigm with implications for breast cancer research.Breast Cancer Res Treat. 1995 Jul;35(1):115-32. doi: 10.1007/BF00694752. Breast Cancer Res Treat. 1995. PMID: 7612898 Review.
-
ErbB Receptors and Cancer.Methods Mol Biol. 2017;1652:3-35. doi: 10.1007/978-1-4939-7219-7_1. Methods Mol Biol. 2017. PMID: 28791631 Review.
Cited by
-
The structural basis for cancer treatment decisions.Oncotarget. 2014 Sep 15;5(17):7285-302. doi: 10.18632/oncotarget.2439. Oncotarget. 2014. PMID: 25277176 Free PMC article. Review.
-
Molecular basis for multimerization in the activation of the epidermal growth factor receptor.Elife. 2016 Mar 28;5:e14107. doi: 10.7554/eLife.14107. Elife. 2016. PMID: 27017828 Free PMC article.
-
Mechanistic Insights into R776H Mediated Activation of Epidermal Growth Factor Receptor Kinase.Biochemistry. 2015 Jul 14;54(27):4216-25. doi: 10.1021/acs.biochem.5b00444. Epub 2015 Jul 6. Biochemistry. 2015. PMID: 26101090 Free PMC article.
-
The juxtamembrane regions of human receptor tyrosine kinases exhibit conserved interaction sites with anionic lipids.Sci Rep. 2015 Mar 17;5:9198. doi: 10.1038/srep09198. Sci Rep. 2015. PMID: 25779975 Free PMC article.
-
Expanding the Disorder-Function Paradigm in the C-Terminal Tails of Erbbs.Biomolecules. 2021 Nov 14;11(11):1690. doi: 10.3390/biom11111690. Biomolecules. 2021. PMID: 34827688 Free PMC article. Review.
References
-
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125: 1137–1149 - PubMed
-
- Margolis BL, Lax I, Kris R, Dombalagian M, Honegger AM, et al. (1989) All autophosphorylation sites of epidermal growth factor receptor and HER2/neu are located in their carboxyl-terminal tails: identification of a novel site in EGF receptor. J Biol Chem 264: 10667–10671 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
