

Midkine and multiple sclerosis
- PMID: 24460675
- PMCID: PMC3925032
- DOI: 10.1111/bph.12499
Midkine and multiple sclerosis
Abstract
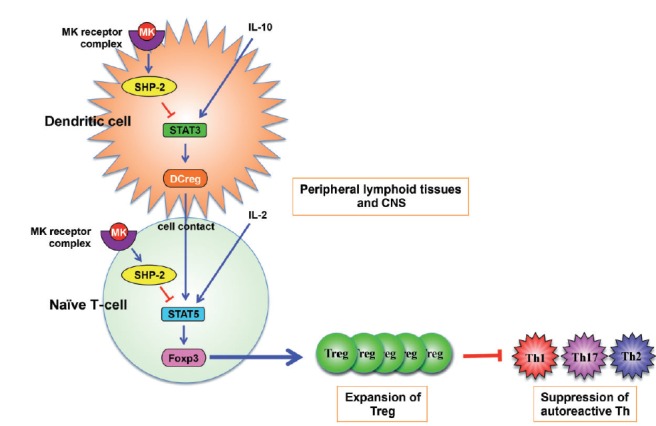
Multiple sclerosis (MS) is an autoimmune neurological disease characterized by inflammatory demyelination with subsequent neuronal damage in the CNS. MS and its animal model, experimental autoimmune encephalomyelitis (EAE), have been thought as autoreactive Th1 and Th17 cell-mediated diseases. CD4(+) CD25(+) FoxP3(+) regulatory T-cell (Treg) plays a pivotal role in autoimmune tolerance, and tolerogenic dendritic cells (DCreg) drive the development of inducible Treg cells. Thus, a dysfunction in the development of Treg and DCreg leads to the development of autoimmune diseases. However, the factors that regulate Treg and DCreg are largely unknown. We recently showed that removal of midkine (MK) suppressed EAE due to an expansion of the Treg cell population as well as a decrease in the numbers of autoreactive Th1 and Th17 cells. MK decreased the Treg cell population by suppressing the phosphorylation of STAT5, which is essential for the expression of Foxp3, the master transcriptional factor of Treg cell differentiation. Furthermore, MK reduces the DCreg cell population by inhibiting the phosphorylation of STAT3, which is critical for DCreg development. Blockade of MK signalling by a specific RNA aptamer significantly elevated the population of DCreg and Treg cells and ameliorated EAE without detectable adverse effects. Therefore, the inhibition of MK may provide an effective therapeutic strategy against autoimmune diseases including MS.
Linked articles: This article is part of a themed section on Midkine. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2014.171.issue-4.
Keywords: aptamer; experimental autoimmune encephalomyelitis; midkine; multiple sclerosis; regulatory T-cell; tolerogenic dendritic cell.
© 2013 The British Pharmacological Society.
Figures
Similar articles
-
Midkine inhibits inducible regulatory T cell differentiation by suppressing the development of tolerogenic dendritic cells.J Immunol. 2012 Mar 15;188(6):2602-11. doi: 10.4049/jimmunol.1102346. Epub 2012 Feb 8. J Immunol. 2012. PMID: 22323540
-
Inhibition of midkine alleviates experimental autoimmune encephalomyelitis through the expansion of regulatory T cell population.Proc Natl Acad Sci U S A. 2008 Mar 11;105(10):3915-20. doi: 10.1073/pnas.0709592105. Epub 2008 Mar 4. Proc Natl Acad Sci U S A. 2008. PMID: 18319343 Free PMC article.
-
Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination.J Neurol Sci. 2013 Oct 15;333(1-2):76-87. doi: 10.1016/j.jns.2013.03.002. Epub 2013 Apr 8. J Neurol Sci. 2013. PMID: 23578791 Free PMC article. Review.
-
Bifidobacterium animalis in combination with human origin of Lactobacillus plantarum ameliorate neuroinflammation in experimental model of multiple sclerosis by altering CD4+ T cell subset balance.Biomed Pharmacother. 2017 Nov;95:1535-1548. doi: 10.1016/j.biopha.2017.08.117. Epub 2017 Sep 22. Biomed Pharmacother. 2017. PMID: 28946394
-
Midkine in nephrogenesis, hypertension and kidney diseases.Br J Pharmacol. 2014 Feb;171(4):879-87. doi: 10.1111/bph.12418. Br J Pharmacol. 2014. PMID: 24106831 Free PMC article. Review.
Cited by
-
An expanded polyglutamine in ATAXIN1 results in a loss-of-function that exacerbates severity of Multiple Sclerosis in an EAE mouse model.Res Sq [Preprint]. 2025 Apr 14:rs.3.rs-5664390. doi: 10.21203/rs.3.rs-5664390/v1. Res Sq. 2025. Update in: J Neuroinflammation. 2025 Apr 30;22(1):127. doi: 10.1186/s12974-025-03450-2. PMID: 40321775 Free PMC article. Updated. Preprint.
-
Sex hormones and neuromyelitis optica spectrum disorder: a bidirectional Mendelian randomization study.Neurol Sci. 2024 Sep;45(9):4471-4479. doi: 10.1007/s10072-024-07501-z. Epub 2024 Apr 2. Neurol Sci. 2024. PMID: 38565746
-
Crosstalk between dendritic cells and regulatory T cells: Protective effect and therapeutic potential in multiple sclerosis.Front Immunol. 2022 Sep 13;13:970508. doi: 10.3389/fimmu.2022.970508. eCollection 2022. Front Immunol. 2022. PMID: 36177043 Free PMC article. Review.
-
Interleukin-19 Abrogates Experimental Autoimmune Encephalomyelitis by Attenuating Antigen-Presenting Cell Activation.Front Immunol. 2021 Mar 11;12:615898. doi: 10.3389/fimmu.2021.615898. eCollection 2021. Front Immunol. 2021. PMID: 33776998 Free PMC article.
-
Differential Production of Midkine and Pleiotrophin by Innate APCs upon Stimulation through Nucleic Acid-Sensing TLRs.J Immunol Res. 2023 Oct 9;2023:7944102. doi: 10.1155/2023/7944102. eCollection 2023. J Immunol Res. 2023. PMID: 37850119 Free PMC article.
References
-
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. - PubMed
-
- Brody EN, Gold L. Aptamers as therapeutic and diagnostic agents. J Biotechnol. 2000;74:5–13. - PubMed
-
- Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
