

ROVER variant caller: read-pair overlap considerate variant-calling software applied to PCR-based massively parallel sequencing datasets
- PMID: 24461215
- PMCID: PMC3904415
- DOI: 10.1186/1751-0473-9-3
ROVER variant caller: read-pair overlap considerate variant-calling software applied to PCR-based massively parallel sequencing datasets
Abstract
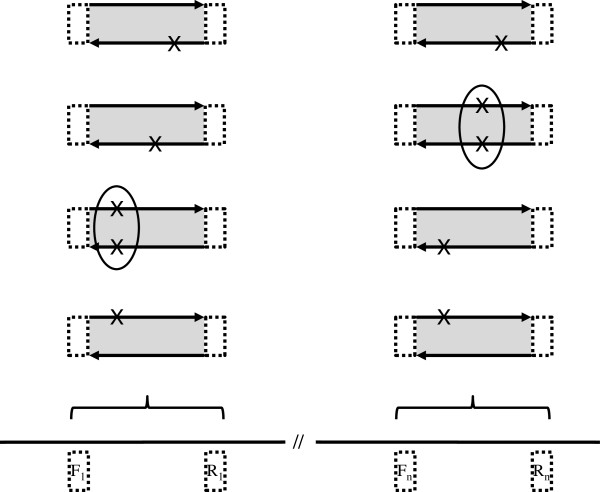
Background: We recently described Hi-Plex, a highly multiplexed PCR-based target-enrichment system for massively parallel sequencing (MPS), which allows the uniform definition of library size so that subsequent paired-end sequencing can achieve complete overlap of read pairs. Variant calling from Hi-Plex-derived datasets can thus rely on the identification of variants appearing in both reads of read-pairs, permitting stringent filtering of sequencing chemistry-induced errors. These principles underly ROVER software (derived from Read Overlap PCR-MPS variant caller), which we have recently used to report the screening for genetic mutations in the breast cancer predisposition gene PALB2. Here, we describe the algorithms underlying ROVER and its usage.
Results: ROVER enables users to quickly and accurately identify genetic variants from PCR-targeted, overlapping paired-end MPS datasets. The open-source availability of the software and threshold tailorability enables broad access for a range of PCR-MPS users.
Methods: ROVER is implemented in Python and runs on all popular POSIX-like operating systems (Linux, OS X). The software accepts a tab-delimited text file listing the coordinates of the target-specific primers used for targeted enrichment based on a specified genome-build. It also accepts aligned sequence files resulting from mapping to the same genome-build. ROVER identifies the amplicon a given read-pair represents and removes the primer sequences by using the mapping co-ordinates and primer co-ordinates. It considers overlapping read-pairs with respect to primer-intervening sequence. Only when a variant is observed in both reads of a read-pair does the signal contribute to a tally of read-pairs containing or not containing the variant. A user-defined threshold informs the minimum number of, and proportion of, read-pairs a variant must be observed in for a 'call' to be made. ROVER also reports the depth of coverage across amplicons to facilitate the identification of any regions that may require further screening.
Conclusions: ROVER can facilitate rapid and accurate genetic variant calling for a broad range of PCR-MPS users.
Figures

References
-
- Nguyen-Dumont T, Pope BJ, Hammet F, Southey MC, Park DJ. A high-plex PCR approach for massively parallel sequencing. Biotechniques. 2013;55:69–74. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
