

Central terminal sensitization of TRPV1 by descending serotonergic facilitation modulates chronic pain
- PMID: 24462040
- PMCID: PMC3943838
- DOI: 10.1016/j.neuron.2013.12.011
Central terminal sensitization of TRPV1 by descending serotonergic facilitation modulates chronic pain
Abstract
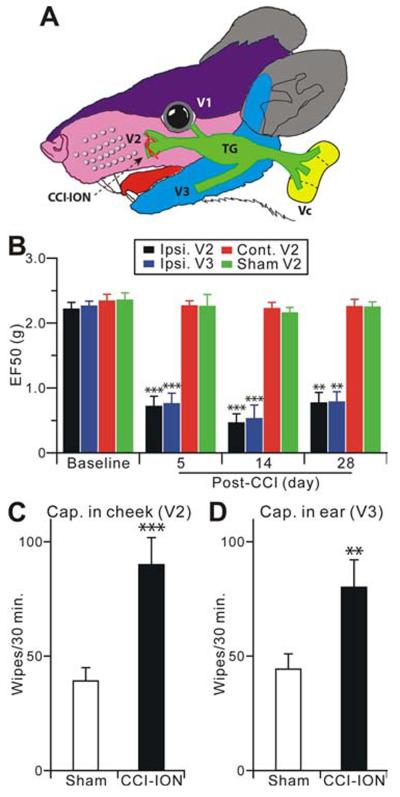
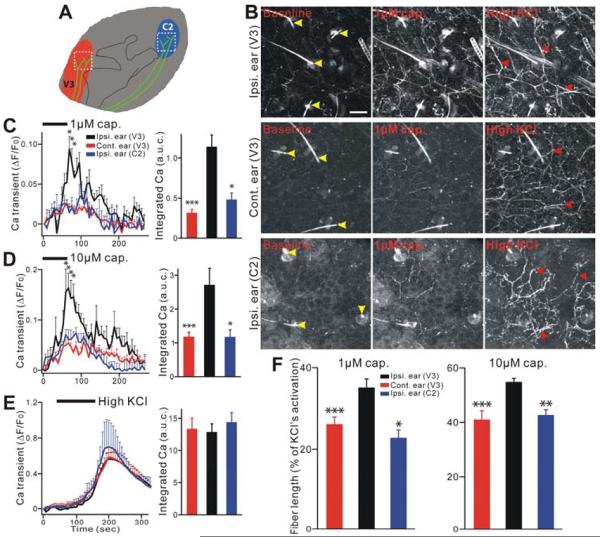
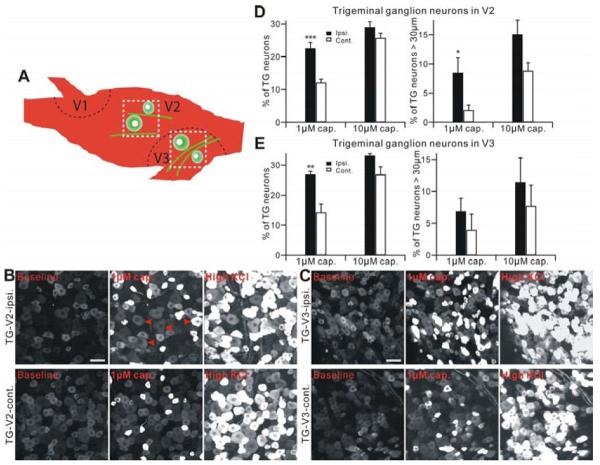
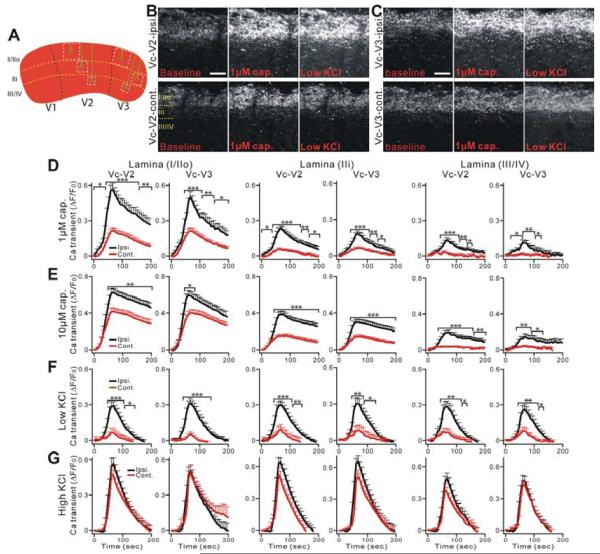
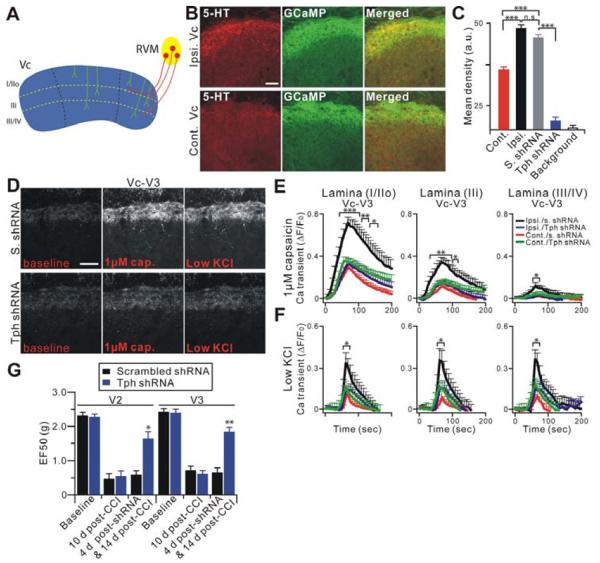
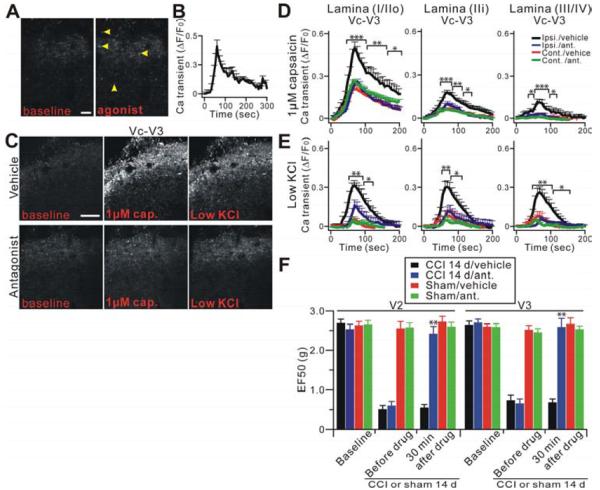
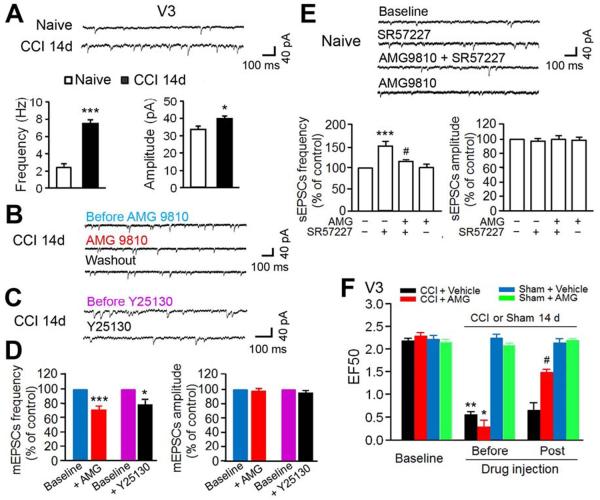
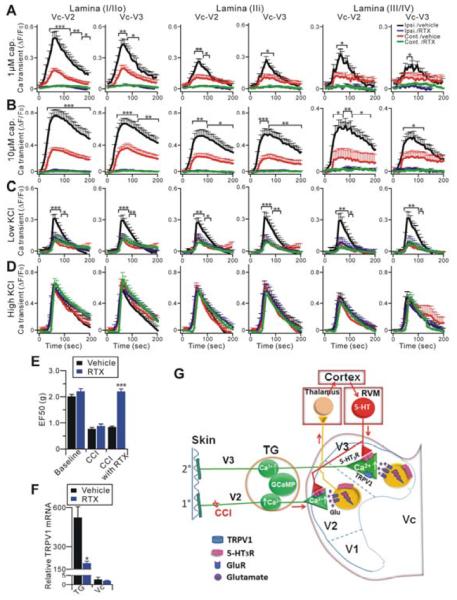
The peripheral terminals of primary nociceptive neurons play an essential role in pain detection mediated by membrane receptors like TRPV1, a molecular sensor of heat and capsaicin. However, the contribution of central terminal TRPV1 in the dorsal horn to chronic pain has not been investigated directly. Combining primary sensory neuron-specific GCaMP3 imaging with a trigeminal neuropathic pain model, we detected robust neuronal hyperactivity in injured and uninjured nerves in the skin, soma in trigeminal ganglion, and central terminals in the spinal trigeminal nucleus. Extensive TRPV1 hyperactivity was observed in central terminals innervating all dorsal horn laminae. The central terminal TRPV1 sensitization was maintained by descending serotonergic (5-HT) input from the brainstem. Central blockade of TRPV1 or 5-HT/5-HT3A receptors attenuated central terminal sensitization, excitatory primary afferent inputs, and mechanical hyperalgesia in the territories of injured and uninjured nerves. Our results reveal central mechanisms facilitating central terminal sensitization underlying chronic pain.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Chronic pain models amplify transient receptor potential vanilloid 1 (TRPV1) receptor responses in adult rat spinal dorsal horn.Neuropharmacology. 2019 Dec 1;160:107753. doi: 10.1016/j.neuropharm.2019.107753. Epub 2019 Sep 4. Neuropharmacology. 2019. PMID: 31493465
-
Stimulating TRPV1 externalization and synthesis in dorsal root ganglion neurons contributes to PGE2 potentiation of TRPV1 activity and nociceptor sensitization.Eur J Pain. 2017 Apr;21(4):575-593. doi: 10.1002/ejp.959. Epub 2016 Oct 14. Eur J Pain. 2017. PMID: 27739618
-
Expression of TRPV1 channels after nerve injury provides an essential delivery tool for neuropathic pain attenuation.PLoS One. 2012;7(9):e44023. doi: 10.1371/journal.pone.0044023. Epub 2012 Sep 4. PLoS One. 2012. PMID: 22962595 Free PMC article.
-
The role of spinal cord vanilloid (TRPV1) receptors in pain modulation.Physiol Res. 2008;57 Suppl 3:S69-S77. doi: 10.33549/physiolres.931601. Epub 2008 May 13. Physiol Res. 2008. PMID: 18481913 Review.
-
Moving towards supraspinal TRPV1 receptors for chronic pain relief.Mol Pain. 2010 Oct 11;6:66. doi: 10.1186/1744-8069-6-66. Mol Pain. 2010. PMID: 20937102 Free PMC article. Review.
Cited by
-
The Secretomes of Painful Versus Nonpainful Human Schwannomatosis Tumor Cells Differentially Influence Sensory Neuron Gene Expression and Sensitivity.Sci Rep. 2019 Sep 11;9(1):13098. doi: 10.1038/s41598-019-49705-w. Sci Rep. 2019. PMID: 31511601 Free PMC article.
-
TRPV1 in Pain and Itch.Adv Exp Med Biol. 2021;1349:249-273. doi: 10.1007/978-981-16-4254-8_12. Adv Exp Med Biol. 2021. PMID: 35138618
-
Botulinum Neurotoxin Chimeras Suppress Stimulation by Capsaicin of Rat Trigeminal Sensory Neurons In Vivo and In Vitro.Toxins (Basel). 2022 Feb 4;14(2):116. doi: 10.3390/toxins14020116. Toxins (Basel). 2022. PMID: 35202143 Free PMC article.
-
Activation of TRPV1 Contributes to Recurrent Febrile Seizures via Inhibiting the Microglial M2 Phenotype in the Immature Brain.Front Cell Neurosci. 2019 Oct 11;13:442. doi: 10.3389/fncel.2019.00442. eCollection 2019. Front Cell Neurosci. 2019. PMID: 31680864 Free PMC article.
-
Imaging the influence of peripheral TRPV1-signaling on cerebral nociceptive processing applying fMRI-based graph theory in a resiniferatoxin rat model.PLoS One. 2022 Apr 28;17(4):e0266669. doi: 10.1371/journal.pone.0266669. eCollection 2022. PLoS One. 2022. PMID: 35482725 Free PMC article.
References
-
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau R.W.t. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. - PubMed
-
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. - PubMed
-
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 NS050274/NS/NINDS NIH HHS/United States
- R01 GM087369/GM/NIGMS NIH HHS/United States
- T32 NS070201/NS/NINDS NIH HHS/United States
- R01GM087369/GM/NIGMS NIH HHS/United States
- R01 NS054791/NS/NINDS NIH HHS/United States
- R01DE022750/DE/NIDCR NIH HHS/United States
- R01 DE022750/DE/NIDCR NIH HHS/United States
- R01 DE021804/DE/NIDCR NIH HHS/United States
- R01DE018573/DE/NIDCR NIH HHS/United States
- K99 NS087088/NS/NINDS NIH HHS/United States
- T32NS070201/NS/NINDS NIH HHS/United States
- R01 DE018573/DE/NIDCR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases