

Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation
- PMID: 24463453
- PMCID: PMC3904626
- DOI: 10.1172/JCI72017
Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation
Abstract
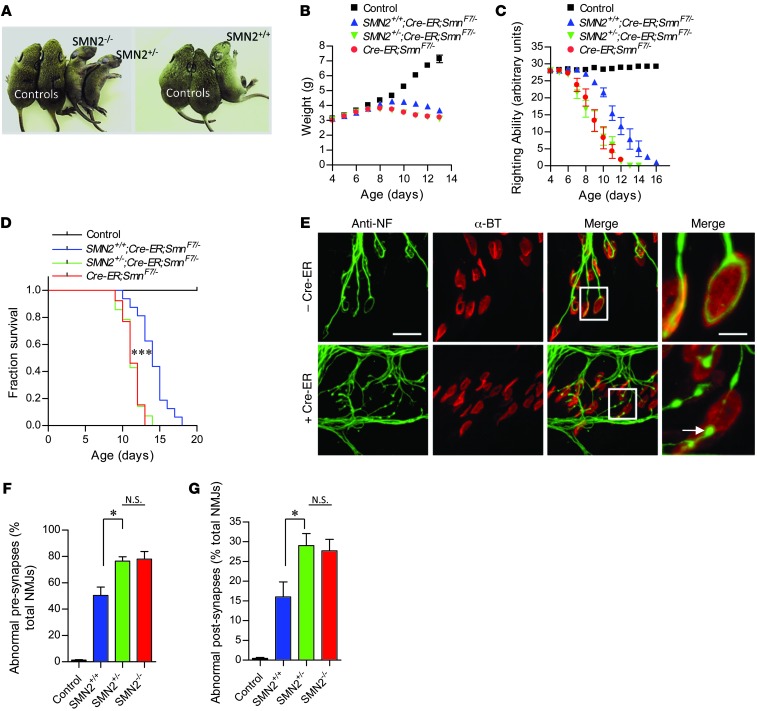
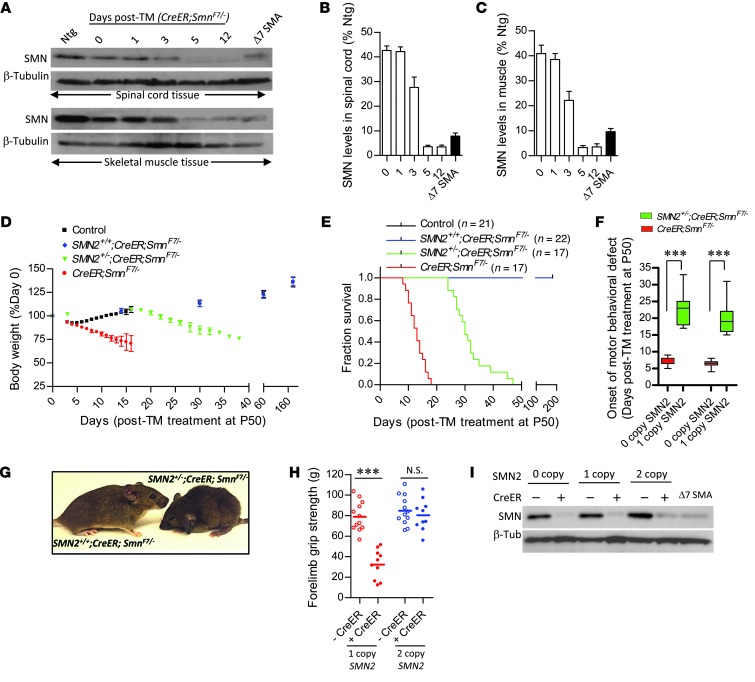
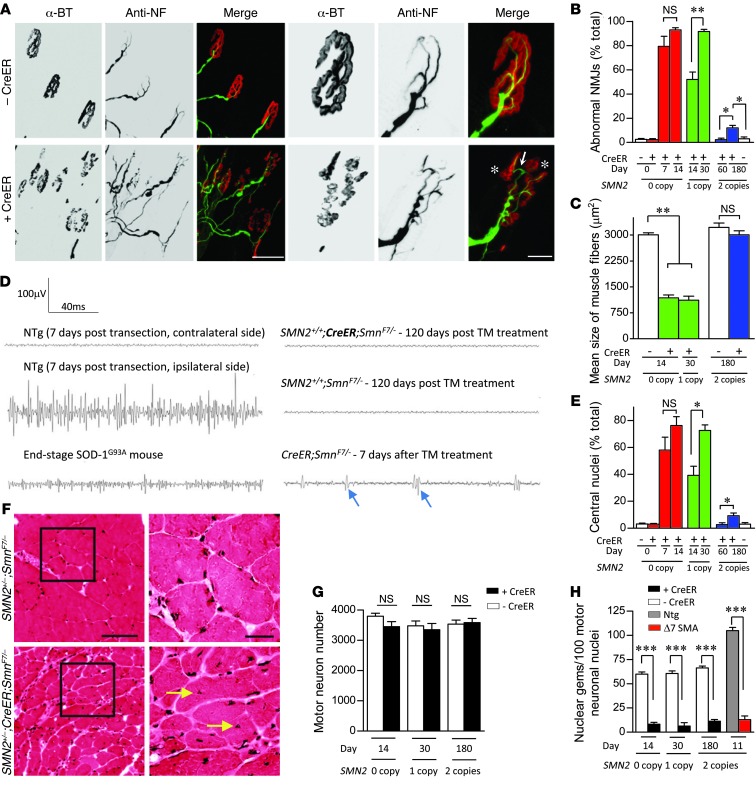
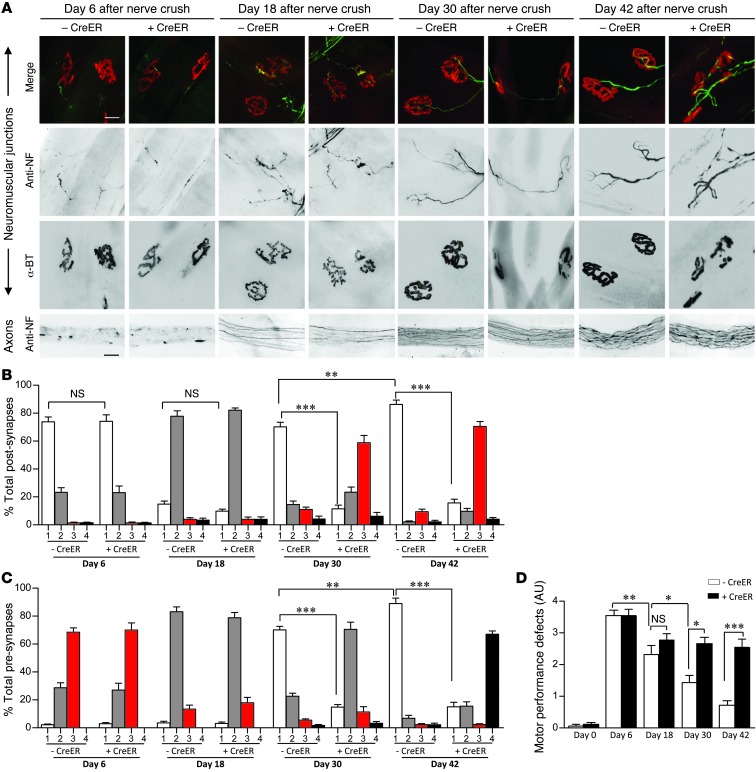
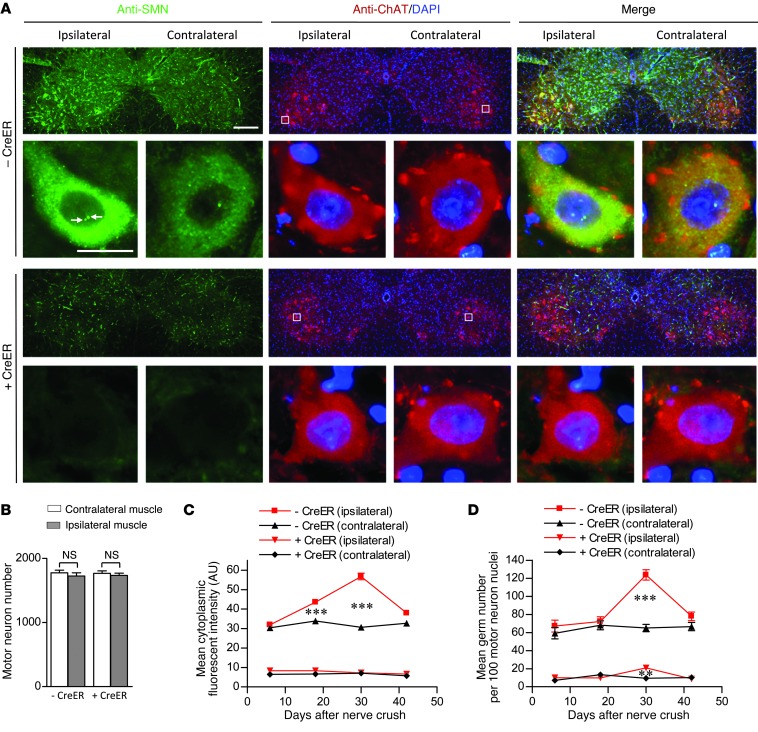
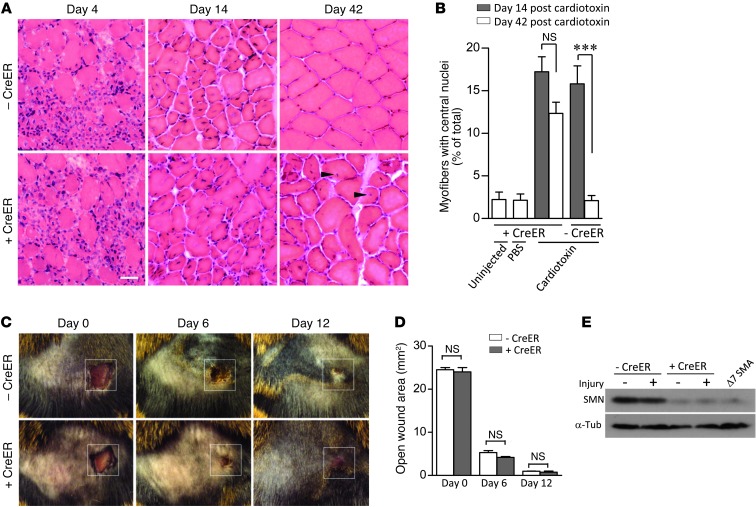
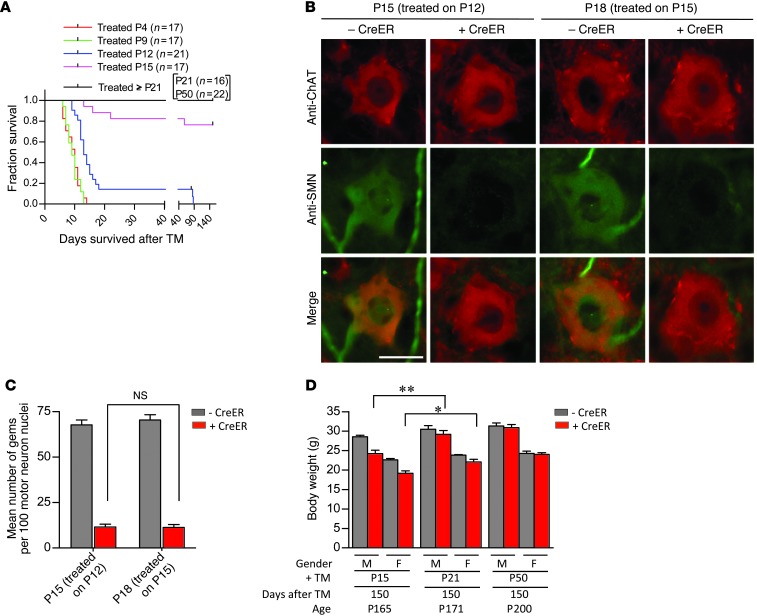
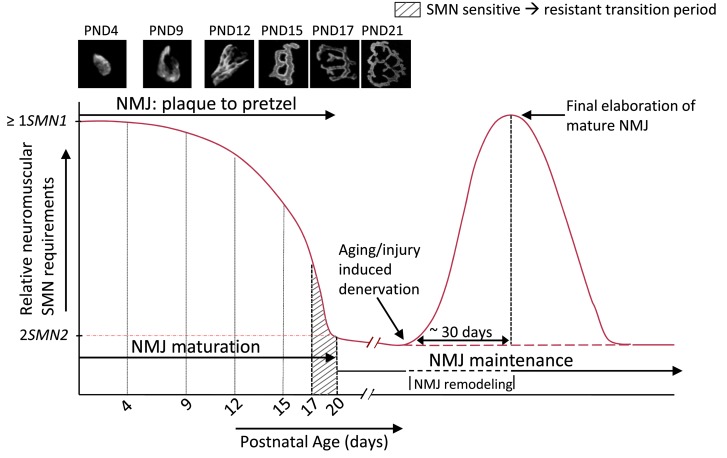
Spinal muscular atrophy is a common motor neuron disease caused by low survival motoneuron (SMN), a key protein in the proper splicing of genes. Restoring the protein is therefore a promising therapeutic strategy. Implementation of this strategy, however, depends on defining the temporal requirements for SMN. Here, we used controlled knockdown of SMN in transgenic mice to determine the precise postnatal stage requirements for this protein. Reducing SMN in neonatal mice resulted in a classic SMA-like phenotype. Unexpectedly, depletion of SMN in adults had relatively little effect. Insensitivity to low SMN emerged abruptly at postnatal day 17, which coincided with establishment of the fully mature neuromuscular junction (NMJ). Mature animals depleted of SMN eventually exhibited evidence of selective neuromuscular pathology that was made worse by traumatic injury. The ability to regenerate the mature NMJ in aged or injured SMN-depleted mice was grossly impaired, a likely consequence of the inability to meet the surge in demand for motoneuronal SMN that was seen in controls. Our results demonstrate that relative maturity of the NMJ determines the temporal requirement for the SMN protein. These observations suggest that the use of potent but potentially deleterious SMN-enhancing agents could be tapered in human patients once the neuromuscular system matures and reintroduced as needed to enhance SMN for remodeling aged or injured NMJs.
Figures
Comment in
-
SMN-targeted therapeutics for spinal muscular atrophy: are we SMArt enough yet?J Clin Invest. 2014 Feb;124(2):487-90. doi: 10.1172/JCI74142. Epub 2014 Jan 27. J Clin Invest. 2014. PMID: 24463455 Free PMC article.
Similar articles
-
Motor neuronal repletion of the NMJ organizer, Agrin, modulates the severity of the spinal muscular atrophy disease phenotype in model mice.Hum Mol Genet. 2017 Jul 1;26(13):2377-2385. doi: 10.1093/hmg/ddx124. Hum Mol Genet. 2017. PMID: 28379354 Free PMC article.
-
Limited phenotypic effects of selectively augmenting the SMN protein in the neurons of a mouse model of severe spinal muscular atrophy.PLoS One. 2012;7(9):e46353. doi: 10.1371/journal.pone.0046353. Epub 2012 Sep 27. PLoS One. 2012. PMID: 23029491 Free PMC article.
-
Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy.J Neurosci. 2012 Jun 20;32(25):8703-15. doi: 10.1523/JNEUROSCI.0204-12.2012. J Neurosci. 2012. PMID: 22723710 Free PMC article.
-
At the "junction" of spinal muscular atrophy pathogenesis: the role of neuromuscular junction dysfunction in SMA disease progression.Curr Mol Med. 2013 Aug;13(7):1160-74. doi: 10.2174/15665240113139990044. Curr Mol Med. 2013. PMID: 23514457 Review.
-
Critical period of neuromuscular development: Importance for a new treatment of SMA.Neuromuscul Disord. 2018 May;28(5):385-393. doi: 10.1016/j.nmd.2018.03.007. Epub 2018 Mar 13. Neuromuscul Disord. 2018. PMID: 29610000 Review.
Cited by
-
Commentary: Amyotrophic Lateral Sclerosis and Myasthenia Gravis Overlap Syndrome: A Review of Two Cases and the Associated Literature.Front Neurol. 2017 Jul 27;8:356. doi: 10.3389/fneur.2017.00356. eCollection 2017. Front Neurol. 2017. PMID: 28798720 Free PMC article. No abstract available.
-
In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy.Brain Sci. 2021 Feb 5;11(2):194. doi: 10.3390/brainsci11020194. Brain Sci. 2021. PMID: 33562482 Free PMC article. Review.
-
Motor neuronal repletion of the NMJ organizer, Agrin, modulates the severity of the spinal muscular atrophy disease phenotype in model mice.Hum Mol Genet. 2017 Jul 1;26(13):2377-2385. doi: 10.1093/hmg/ddx124. Hum Mol Genet. 2017. PMID: 28379354 Free PMC article.
-
AAV9-Stathmin1 gene delivery improves disease phenotype in an intermediate mouse model of spinal muscular atrophy.Hum Mol Genet. 2019 Nov 15;28(22):3742-3754. doi: 10.1093/hmg/ddz188. Hum Mol Genet. 2019. PMID: 31363739 Free PMC article.
-
SMN Protein Can Be Reliably Measured in Whole Blood with an Electrochemiluminescence (ECL) Immunoassay: Implications for Clinical Trials.PLoS One. 2016 Mar 8;11(3):e0150640. doi: 10.1371/journal.pone.0150640. eCollection 2016. PLoS One. 2016. PMID: 26953792 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
