

A polygenic burden of rare disruptive mutations in schizophrenia
- PMID: 24463508
- PMCID: PMC4136494
- DOI: 10.1038/nature12975
A polygenic burden of rare disruptive mutations in schizophrenia
Abstract
Schizophrenia is a common disease with a complex aetiology, probably involving multiple and heterogeneous genetic factors. Here, by analysing the exome sequences of 2,536 schizophrenia cases and 2,543 controls, we demonstrate a polygenic burden primarily arising from rare (less than 1 in 10,000), disruptive mutations distributed across many genes. Particularly enriched gene sets include the voltage-gated calcium ion channel and the signalling complex formed by the activity-regulated cytoskeleton-associated scaffold protein (ARC) of the postsynaptic density, sets previously implicated by genome-wide association and copy-number variation studies. Similar to reports in autism, targets of the fragile X mental retardation protein (FMRP, product of FMR1) are enriched for case mutations. No individual gene-based test achieves significance after correction for multiple testing and we do not detect any alleles of moderately low frequency (approximately 0.5 to 1 per cent) and moderately large effect. Taken together, these data suggest that population-based exome sequencing can discover risk alleles and complements established gene-mapping paradigms in neuropsychiatric disease.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
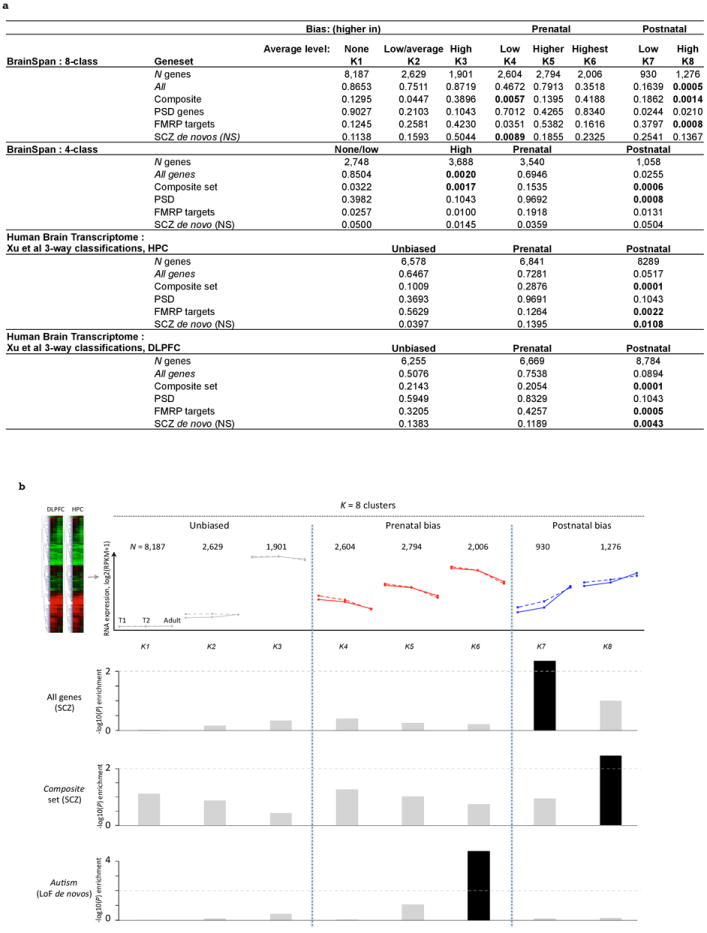
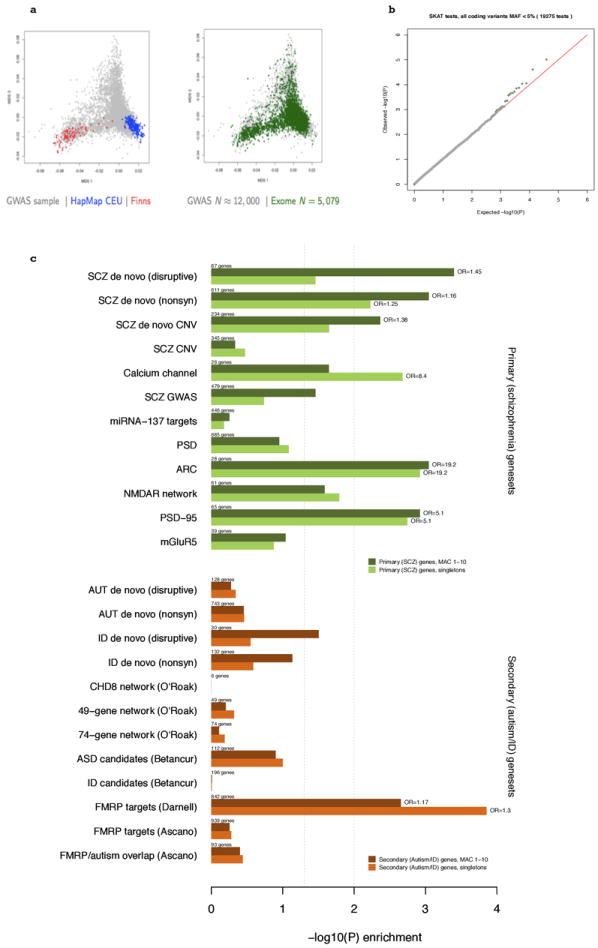
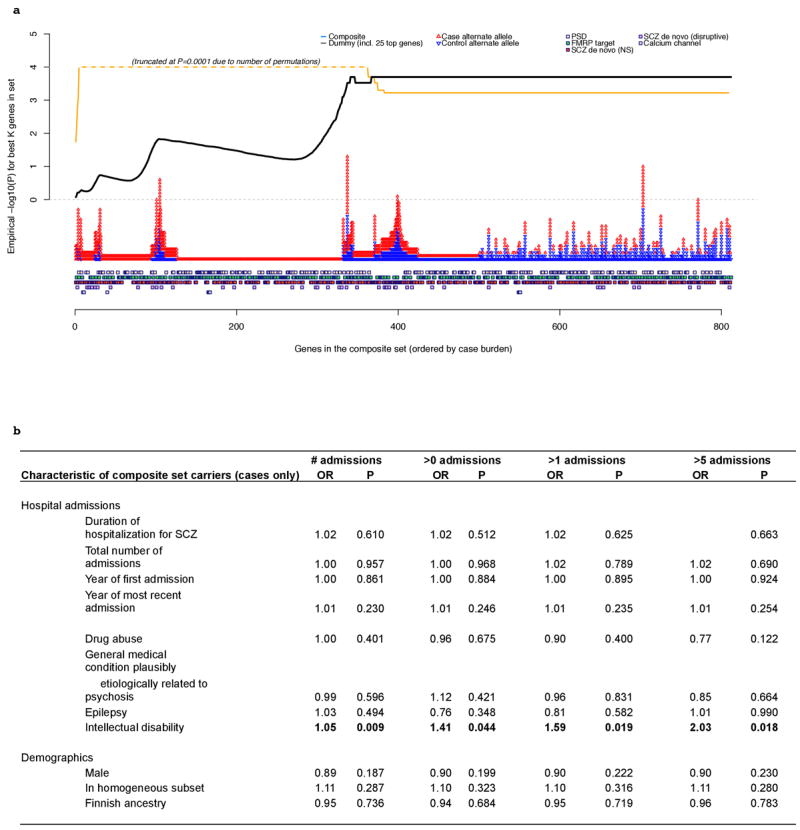
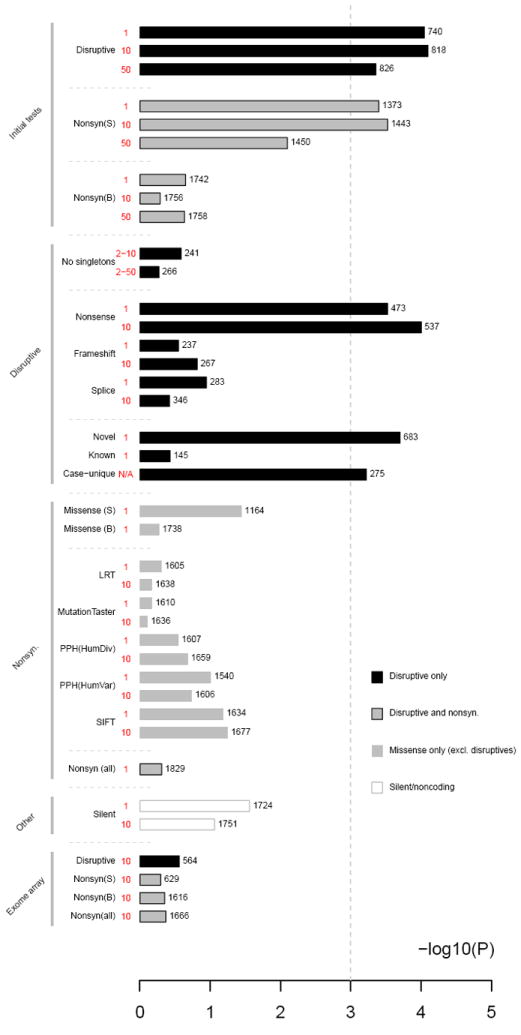
Comment in
-
Genetics: finding genes for schizophrenia.Curr Biol. 2014 Aug 18;24(16):R755-7. doi: 10.1016/j.cub.2014.07.018. Curr Biol. 2014. PMID: 25137590 Free PMC article.
References
-
- Albrechtsen A, Grarup N, Li Y, Sparsø T, Tian G, Cao H, Jiang T, Kim SY, Korneliussen T, Li Q, Nie C, Wu R, Skotte L, Morris AP, Ladenvall C, Cauchi S, Stančáková A, Andersen G, Astrup A, Banasik K, Bennett AJ, Bolund L, Charpentier G, Chen Y, Dekker JM, Doney AS, Dorkhan M, Forsen T, Frayling TM, Groves CJ, Gui Y, Hallmans G, Hattersley AT, He K, Hitman GA, Holmkvist J, Huang S, Jiang H, Jin X, Justesen JM, Kristiansen K, Kuusisto J, Lajer M, Lantieri O, Li W, Liang H, Liao Q, Liu X, Ma T, Ma X, Manijak MP, Marre M, Mokrosiński J, Morris AD, Mu B, Nielsen AA, Nijpels G, Nilsson P, Palmer CN, Rayner NW, Renström F, Ribel-Madsen R, Robertson N, Rolandsson O, Rossing P, Schwartz TW, Slagboom PE, Sterner M, Tang M, Tarnow L, Tuomi T, Van’t Riet E, van Leeuwen N, Varga TV, Vestmar MA, Walker M, Wang B, Wang Y, Wu H, Xi F, Yengo L, Yu C, Zhang X, Zhang J, Zhang Q, Zhang W, Zheng H, Zhou Y, Altshuler D, ‘t Hart LM, Franks PW, Balkau B, Froguel P, McCarthy MI, Laakso M, Groop L, Christensen C, Brandslund I, Lauritzen T, Witte DR, Linneberg A, Jørgensen T, Hansen T, Wang J, Nielsen R, Pedersen O D.E.S.I.R. Study Group, the DIAGRAM Consortium. Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia. 2013;56(2):298–310. - PMC - PubMed
-
- Berryer MH, Hamdan FF, Klitten LL, Møller RS, Carmant L, Schwartzentruber J, Patry L, Dobrzeniecka S, Rochefort D, Neugnot-Cerioli M, Lacaille JC, Niu Z, Eng CM, Yang Y, Palardy S, Belhumeur C, Rouleau GA, Tommerup N, Immken L, Beauchamp MH, Patel GS, Majewski J, Tarnopolsky MA, Scheffzek K, Hjalgrim H, Michaud JL, Di Cristo G. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat. 2013;34(2):385–94. - PubMed
-
- Callan MA, Zarnescu DC. Heads-up: new roles for the fragile × mental retardation protein in neural stem and progenitor cells. Genesis. 2011 Jun;49(6):424–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 MH095034/MH/NIMH NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- R01 MH091115/MH/NIMH NIH HHS/United States
- TT32MH017119/MH/NIMH NIH HHS/United States
- U54HG003067/HG/NHGRI NIH HHS/United States
- R01 MH099126/MH/NIMH NIH HHS/United States
- G0802238/MRC_/Medical Research Council/United Kingdom
- T32 MH017119/MH/NIMH NIH HHS/United States
- R01 MH095088/MH/NIMH NIH HHS/United States
- RC2 MH089905/MH/NIMH NIH HHS/United States
- R01 MH077139/MH/NIMH NIH HHS/United States
- I01 BX002395/BX/BLRD VA/United States
- RC2MH089905/MH/NIMH NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 HG005827/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
