

Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis
- PMID: 24463883
- PMCID: PMC4030775
- DOI: 10.1093/hmg/ddu030
Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis
Abstract
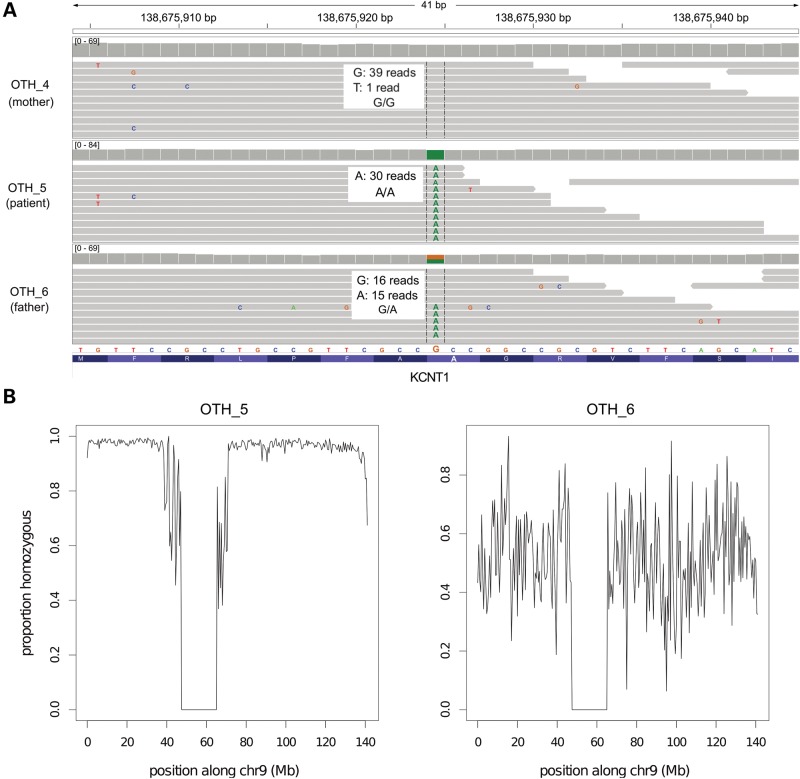
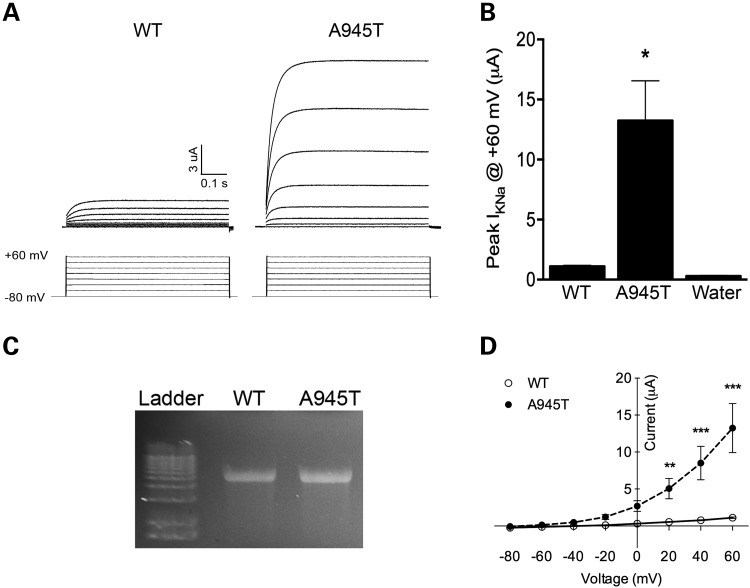
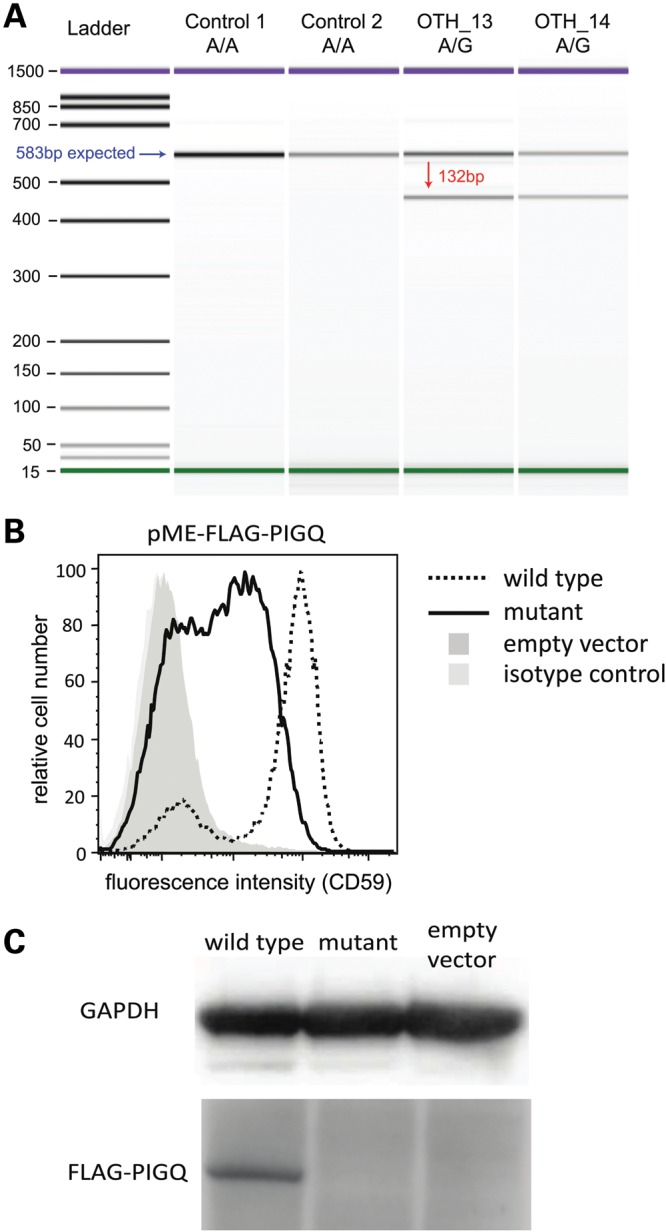
In severe early-onset epilepsy, precise clinical and molecular genetic diagnosis is complex, as many metabolic and electro-physiological processes have been implicated in disease causation. The clinical phenotypes share many features such as complex seizure types and developmental delay. Molecular diagnosis has historically been confined to sequential testing of candidate genes known to be associated with specific sub-phenotypes, but the diagnostic yield of this approach can be low. We conducted whole-genome sequencing (WGS) on six patients with severe early-onset epilepsy who had previously been refractory to molecular diagnosis, and their parents. Four of these patients had a clinical diagnosis of Ohtahara Syndrome (OS) and two patients had severe non-syndromic early-onset epilepsy (NSEOE). In two OS cases, we found de novo non-synonymous mutations in the genes KCNQ2 and SCN2A. In a third OS case, WGS revealed paternal isodisomy for chromosome 9, leading to identification of the causal homozygous missense variant in KCNT1, which produced a substantial increase in potassium channel current. The fourth OS patient had a recessive mutation in PIGQ that led to exon skipping and defective glycophosphatidyl inositol biosynthesis. The two patients with NSEOE had likely pathogenic de novo mutations in CBL and CSNK1G1, respectively. Mutations in these genes were not found among 500 additional individuals with epilepsy. This work reveals two novel genes for OS, KCNT1 and PIGQ. It also uncovers unexpected genetic mechanisms and emphasizes the power of WGS as a clinical tool for making molecular diagnoses, particularly for highly heterogeneous disorders.
© The Author 2014. Published by Oxford University Press.
Figures
References
-
- Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. - PubMed
-
- Rabbani B., Mahdieh N., Hosomichi K., Nakaoka H., Inoue I. Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J. Hum. Genet. 2012;57:621–632. - PubMed
-
- Sharma V.P., Fenwick A.L., Brockop M.S., McGowan S.J., Goos J.A., Hoogeboom A.J., Brady A.F., Jeelani N.O., Lynch S.A., Mulliken J.B., et al. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat. Genet. 2013;45:304–307. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
