

Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation
- PMID: 24464042
- PMCID: PMC3937121
- DOI: 10.1038/nn.3637
Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation
Abstract
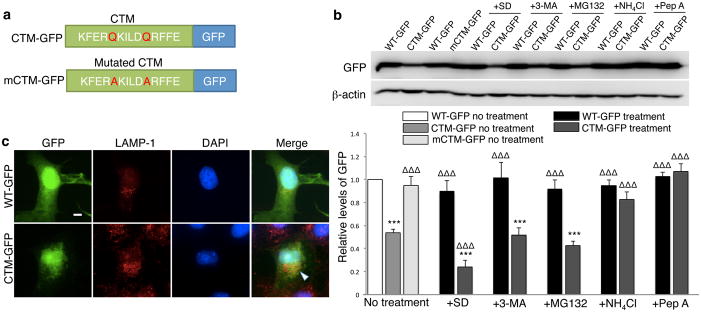
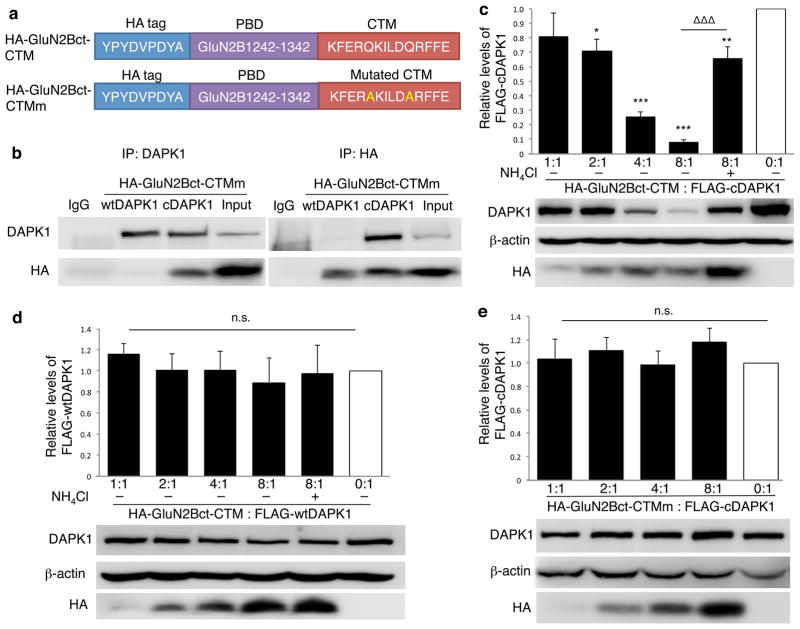
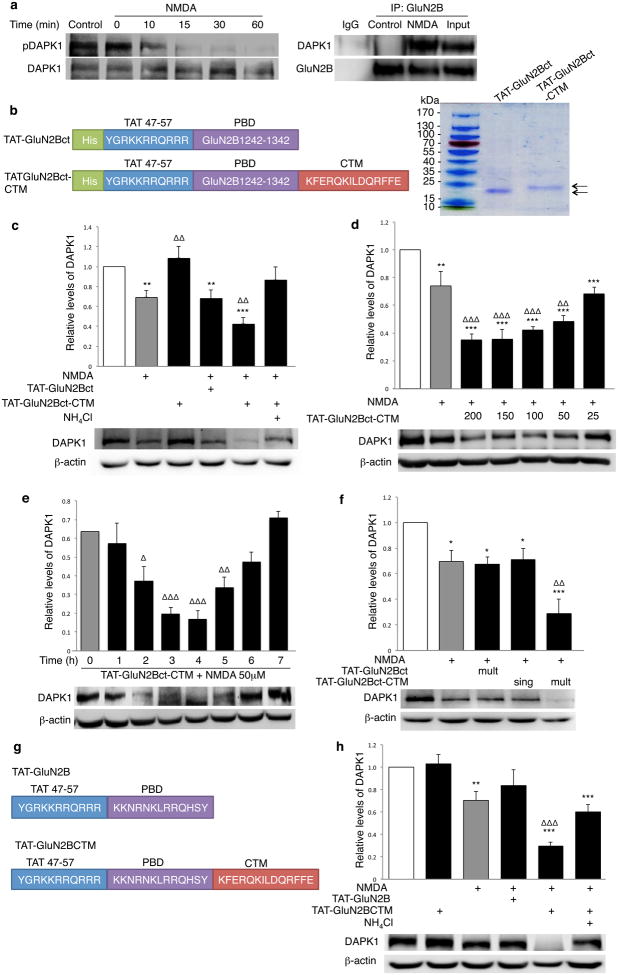
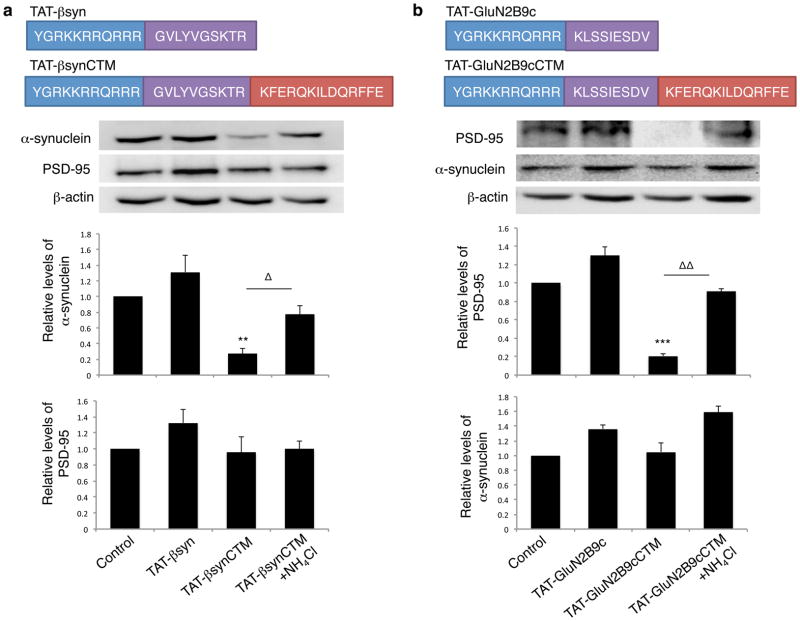
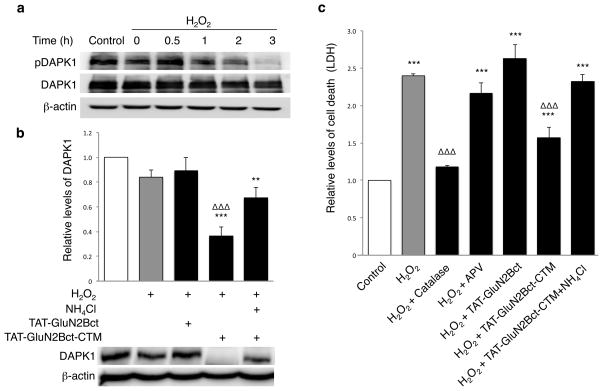
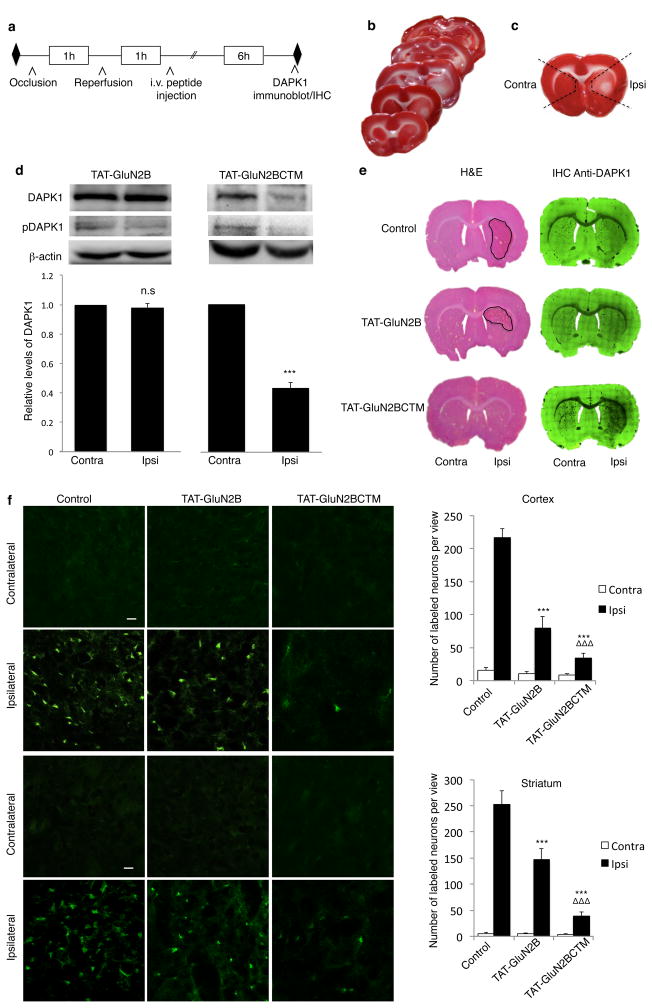
Rapid and reversible methods for altering the levels of endogenous proteins are critically important for studying biological systems and developing therapeutics. Here we describe a membrane-permeant targeting peptide-based method that rapidly and reversibly knocks down endogenous proteins through chaperone-mediated autophagy in vitro and in vivo. We demonstrate the specificity, efficacy and generalizability of the method by showing efficient knockdown of various proteins, including death associated protein kinase 1 (160 kDa), scaffolding protein PSD-95 (95 kDa) and α-synuclein (18 kDa), with their respective targeting peptides in a dose-, time- and lysosomal activity-dependent manner in rat neuronal cultures. Moreover, we show that, when given systemically, the peptide system efficiently knocked down the targeted protein in the brains of intact rats. Our study provides a robust and convenient research tool for manipulating endogenous protein levels and may also lead to the development of protein knockdown-based therapeutics for treating human diseases.
Figures
Comment in
-
Knocking down Goliath.Nat Methods. 2014 Mar;11(3):232. doi: 10.1038/nmeth.2875. Nat Methods. 2014. PMID: 24724170 No abstract available.
References
-
- Houdebine LM. Transgenic animal models in biomedical research. Methods Mol Biol. 2007;360:163–202. - PubMed
-
- Yamamoto A, Hen R, Dauer WT. The Ons and Offs of Inducible Transgenic Technology: A Review. Neurobiology of Disease. 2001;8:923–932. - PubMed
-
- Banaszynski LA, Wandless TJ. Conditional Control of Protein Function. Chemistry & Biology. 2006;13:11–21. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
