

Contractile dysfunction in a mouse model expressing a heterozygous MYBPC3 mutation associated with hypertrophic cardiomyopathy
- PMID: 24464755
- PMCID: PMC3949045
- DOI: 10.1152/ajpheart.00913.2013
Contractile dysfunction in a mouse model expressing a heterozygous MYBPC3 mutation associated with hypertrophic cardiomyopathy
Abstract
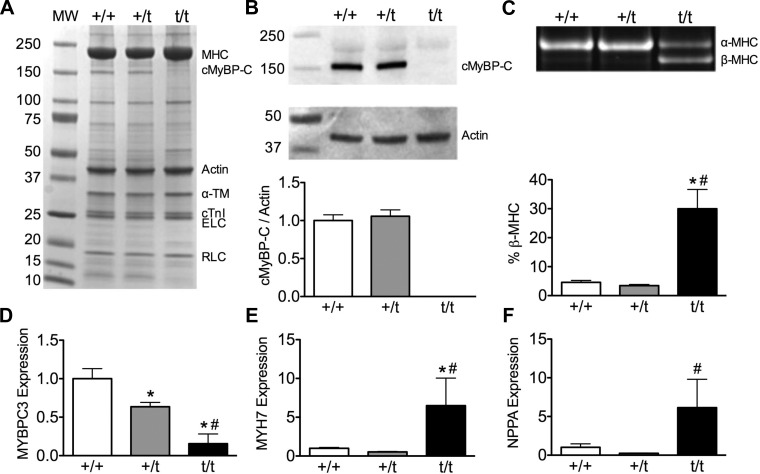
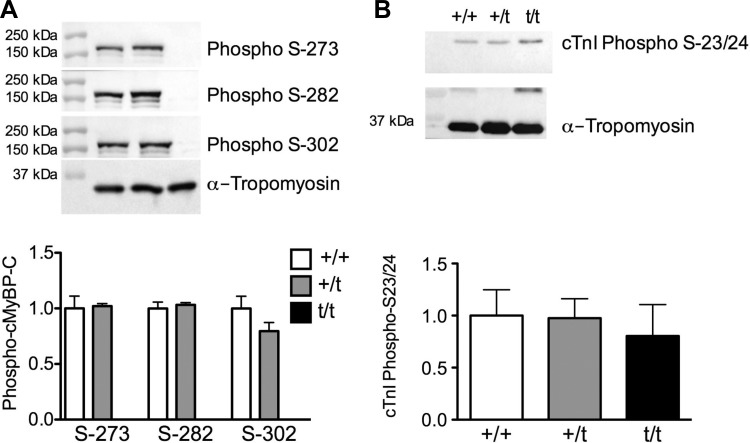
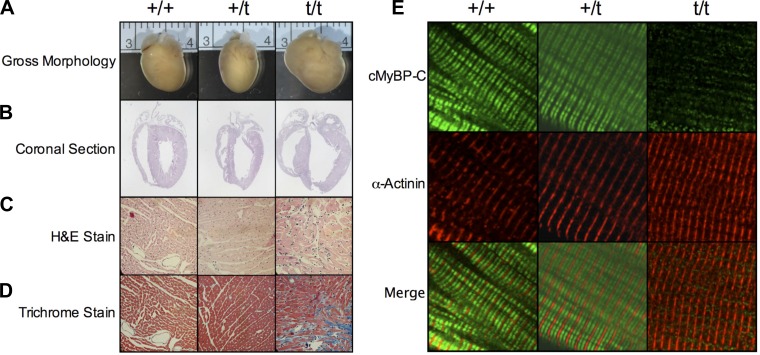
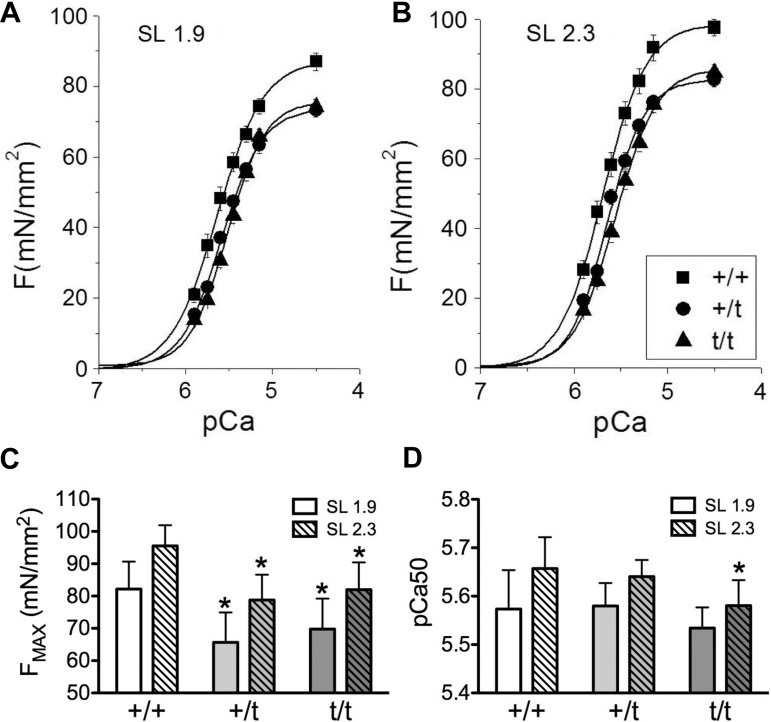
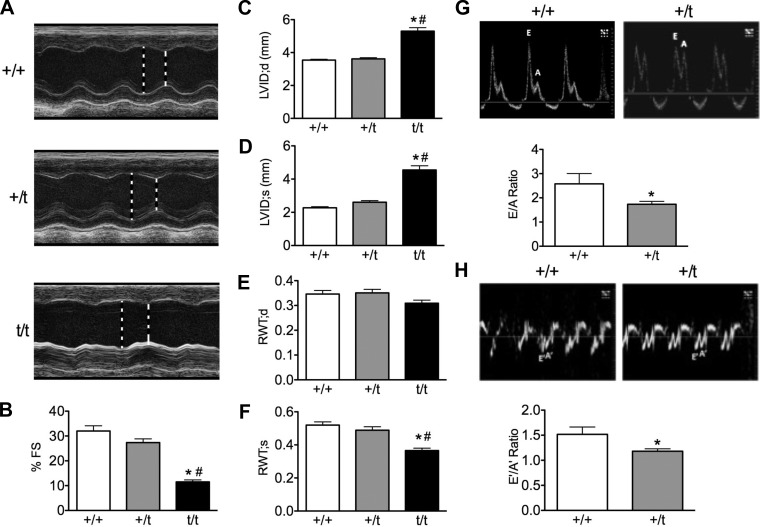
The etiology of hypertrophic cardiomyopathy (HCM) has been ascribed to mutations in genes encoding sarcomere proteins. In particular, mutations in MYBPC3, a gene which encodes cardiac myosin binding protein-C (cMyBP-C), have been implicated in over one third of HCM cases. Of these mutations, 70% are predicted to result in C'-truncated protein products, which are undetectable in tissue samples. Heterozygous carriers of these truncation mutations exhibit varying penetrance of HCM, with symptoms often occurring later in life. We hypothesize that heterozygous carriers of MYBPC3 mutations, while seemingly asymptomatic, have subtle functional impairments that precede the development of overt HCM. This study compared heterozygous (+/t) knock-in MYBPC3 truncation mutation mice with wild-type (+/+) littermates to determine whether functional alterations occur at the whole-heart or single-cell level before the onset of hypertrophy. The +/t mice show ∼40% reduction in MYBPC3 transcription, but no changes in cMyBP-C level, phosphorylation status, or cardiac morphology. Nonetheless, +/t mice show significantly decreased maximal force development at sarcomere lengths of 1.9 μm (+/t 68.5 ± 4.1 mN/mm(2) vs. +/+ 82.2 ± 3.2) and 2.3 μm (+/t 79.2 ± 3.1 mN/mm(2) vs. +/+ 95.5 ± 2.4). In addition, heterozygous mice show significant reductions in vivo in the early/after (E/A) (+/t 1.74 ± 0.12 vs. +/+ 2.58 ± 0.43) and E'/A' (+/t 1.18 ± 0.05 vs. +/+ 1.52 ± 0.15) ratios, indicating diastolic dysfunction. These results suggest that seemingly asymptomatic heterozygous MYBPC3 carriers do suffer impairments that may presage the onset of HCM.
Keywords: cardiac myosin binding protein-C; haploinsufficiency.
Figures
References
-
- Alcalai R, Seidman J, Seidman C. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol 19: 104–110, 2008 - PubMed
-
- Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, Gautel M, Labeit S, James M, Beckmann J, Weissenbach J, Vosberg HP, Fiszman M, Komajda M, Schwartz K. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet 11: 438–440, 1995 - PubMed
-
- Carrier L, Bonne G, Bahrend E, Yu B, Richard P, Niel F, Hainque B, Cruaud C, Gary F, Labeit S, Bouhour J, Dubourg O, Desnos M, Hagege A, Trent R, Komajda M, Fiszman M, Schwartz K. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ Res 80: 427–434, 1997 - PubMed
-
- Carrier L, Knoll R, Vignier N, Keller D, Bausero P, Prudhon B, Isnard R, Ambroisine M, Fiszman M, Ross J, Schwartz K, Chien K. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc Res 63: 293–304, 2004 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases