

Integrative genetic characterization and phenotype correlations in pheochromocytoma and paraganglioma tumours
- PMID: 24466223
- PMCID: PMC3899286
- DOI: 10.1371/journal.pone.0086756
Integrative genetic characterization and phenotype correlations in pheochromocytoma and paraganglioma tumours
Abstract
Background: About 60% of Pheochromocytoma (PCC) and Paraganglioma (PGL) patients have either germline or somatic mutations in one of the 12 proposed disease causing genes; SDHA, SDHB, SDHC, SDHD, SDHAF2, VHL, EPAS1, RET, NF1, TMEM127, MAX and H-RAS. Selective screening for germline mutations is routinely performed in clinical management of these diseases. Testing for somatic alterations is not performed on a regular basis because of limitations in interpreting the results.
Aim: The purpose of the study was to investigate genetic events and phenotype correlations in a large cohort of PCC and PGL tumours.
Methods: A total of 101 tumours from 89 patients with PCC and PGL were re-sequenced for a panel of 10 disease causing genes using automated Sanger sequencing. Selected samples were analysed with Multiplex Ligation-dependent Probe Amplification and/or SNParray.
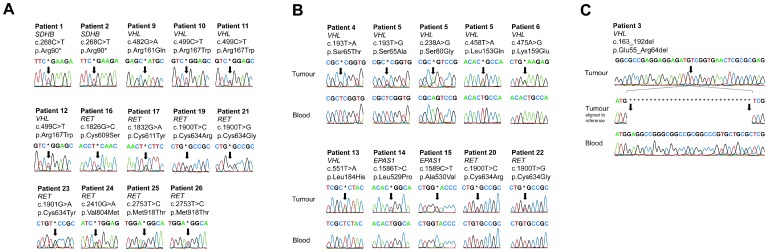
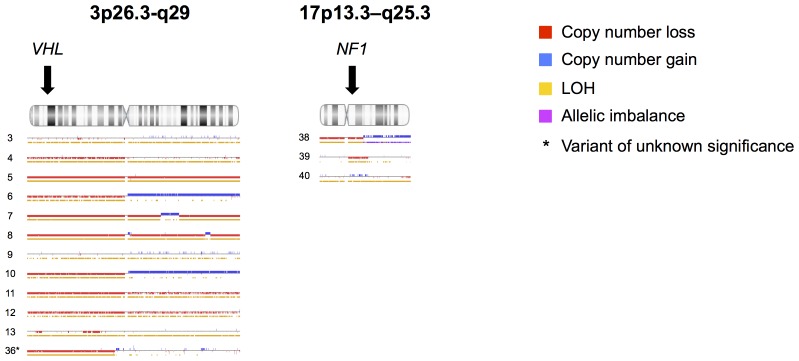
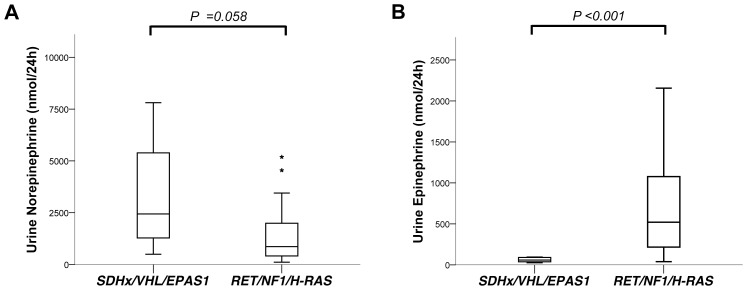
Results: Pathogenic genetic variants were found in tumours from 33 individual patients (37%), 14 (16%) were discovered in constitutional DNA and 16 (18%) were confirmed as somatic. Loss of heterozygosity (LOH) was observed in 1/1 SDHB, 11/11 VHL and 3/3 NF1-associated tumours. In patients with somatic mutations there were no recurrences in contrast to carriers of germline mutations (P = 0.022). SDHx/VHL/EPAS1 associated cases had higher norepinephrine output (P = 0.03) and lower epinephrine output (P<0.001) compared to RET/NF1/H-RAS cases.
Conclusion: Somatic mutations are frequent events in PCC and PGL tumours. Tumour genotype may be further investigated as prognostic factors in these diseases. Growing evidence suggest that analysis of tumour DNA could have an impact on the management of these patients.
Conflict of interest statement
Figures
Similar articles
-
Germline mutations and genotype-phenotype correlation in Asian Indian patients with pheochromocytoma and paraganglioma.Eur J Endocrinol. 2016 Oct;175(4):311-23. doi: 10.1530/EJE-16-0126. Eur J Endocrinol. 2016. PMID: 27539324
-
MAX mutations status in Swedish patients with pheochromocytoma and paraganglioma tumours.Fam Cancer. 2014 Mar;13(1):121-5. doi: 10.1007/s10689-013-9666-3. Fam Cancer. 2014. PMID: 23743562
-
Novel SDHB and TMEM127 Mutations in Patients with Pheochromocytoma/Paraganglioma Syndrome.Pathol Oncol Res. 2016 Oct;22(4):673-9. doi: 10.1007/s12253-016-0050-0. Epub 2016 Mar 9. Pathol Oncol Res. 2016. PMID: 26960314
-
An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma.Best Pract Res Clin Endocrinol Metab. 2020 Mar;34(2):101416. doi: 10.1016/j.beem.2020.101416. Epub 2020 Mar 10. Best Pract Res Clin Endocrinol Metab. 2020. PMID: 32295730 Review.
-
Risk of metastatic pheochromocytoma and paraganglioma in SDHx mutation carriers: a systematic review and updated meta-analysis.J Med Genet. 2020 Apr;57(4):217-225. doi: 10.1136/jmedgenet-2019-106324. Epub 2019 Oct 24. J Med Genet. 2020. PMID: 31649053
Cited by
-
Metastatic cluster 2-related pheochromocytoma/paraganglioma: a single-center experience and systematic review.Endocr Connect. 2021 Nov 11;10(11):1463-1476. doi: 10.1530/EC-21-0455. Endocr Connect. 2021. PMID: 34662294 Free PMC article.
-
A meta-analysis of different von Hippel Lindau mutations: are they related to retinal capillary hemangioblastoma?Mol Genet Genomics. 2022 Nov;297(6):1615-1626. doi: 10.1007/s00438-022-01940-z. Epub 2022 Aug 25. Mol Genet Genomics. 2022. PMID: 36006455
-
Absence of BRAF mutation in pheochromocytoma and paraganglioma.Neoplasma. 2017;64(2):278-282. doi: 10.4149/neo_2017_215. Neoplasma. 2017. PMID: 28043156 Free PMC article.
-
New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification.Endocr Rev. 2017 Dec 1;38(6):489-515. doi: 10.1210/er.2017-00062. Endocr Rev. 2017. PMID: 28938417 Free PMC article. Review.
-
Intricacies of the Molecular Machinery of Catecholamine Biosynthesis and Secretion by Chromaffin Cells of the Normal Adrenal Medulla and in Pheochromocytoma and Paraganglioma.Cancers (Basel). 2019 Aug 6;11(8):1121. doi: 10.3390/cancers11081121. Cancers (Basel). 2019. PMID: 31390824 Free PMC article. Review.
References
-
- Mannelli M, Castellano M, Schiavi F, Filetti S, Giacche M, et al. (2009) Clinically guided genetic screening in a large cohort of italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab 94: 1541–1547. - PubMed
-
- Amar L, Servais A, Gimenez-Roqueplo AP, Zinzindohoue F, Chatellier G, et al... (2005) Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab 90: 2110–2116. Epub 2005 Jan 2111. - PubMed
-
- Darr R, Lenders JW, Hofbauer LC, Naumann B, Bornstein SR, et al. (2012) Pheochromocytoma - update on disease management. Ther Adv Endocrinol Metab 3: 11–26 doi: 10.1177/2042018812437356 - DOI - PMC - PubMed
-
- Scott HW Jr, Halter SA (1984) Oncologic aspects of pheochromocytoma: the importance of follow-up. Surgery 96: 1061–1066. - PubMed
-
- Van Slycke S, Caiazzo R, Pigny P, Cardot-Bauters C, Arnalsteen L, et al. (2009) Local-regional recurrence of sporadic or syndromic abdominal extra-adrenal paraganglioma: incidence, characteristics, and outcome. Surgery 146: 986–992 doi: 910.1016/j.surg.2009.1010.1055 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
