

Review
doi: 10.1186/gb4152.
Methods for comprehensive experimental identification of RNA-protein interactions
- PMID: 24467948
- PMCID: PMC4054858
- DOI: 10.1186/gb4152
Item in Clipboard
Review
Methods for comprehensive experimental identification of RNA-protein interactions
Genome Biol.
.
Abstract
The importance of RNA-protein interactions in controlling mRNA regulation and non-coding RNA function is increasingly appreciated. A variety of methods exist to comprehensively define RNA-protein interactions. We describe these methods and the considerations required for designing and interpreting these experiments.
Figures
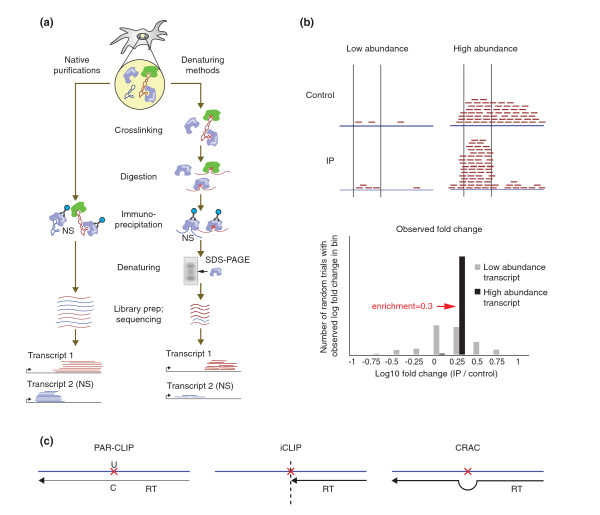
Protein-centric methods for detecting RNA-protein interactions. (a) Schematic of native and denaturing methods. RNA-protein crosslinks are represented by red Xs. Non-specific interactions in solution are labeled (NS) and represented by blue RNA fragments. (b) Computational considerations for identification of interaction sites. The top panel depicts two transcripts - one low-abundance and one high-abundance - that both contain a region that is twofold enriched in the immunoprecipitated (IP) sample over a control. Enrichment measurements in the low-abundance case suffer from high variance. The bottom panel shows simulated enrichment values in a low-abundance region and a high-abundance region, which both have a twofold enrichment in the IP sample. For the low abundance region, the observed log-fold changes are often far from the true underlying value while the abundant transcript shows a more consistent enrichment estimation. (c) A schematic of methods for mapping the precise protein binding sites on RNA. PAR-CLIP takes advantage of U → C transitions induced by UV crosslinking after 4SU incorporation. iCLIP uses the occasional arrest of reverse transcription at crosslink sites and tags and sequences these positions. CRAC relies on reverse transcription errors (deletions and substitutions) at crosslink sites to map sites. CRAC, cross-linking and analysis of cDNA; iCLIP, individual-nucleotide resolution cross-linking and immunoprecipitation; PAR-CLIP, photoactivatable-ribonucleoside-enhanced cross-linking and immunoprecipitation.
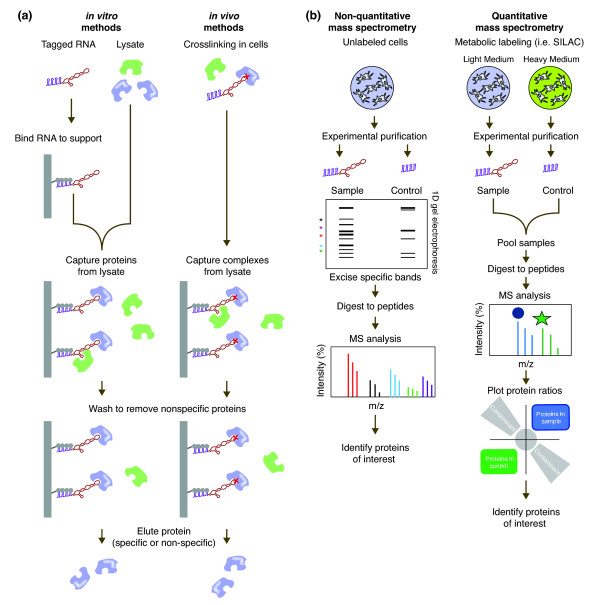
RNA-centric methods for the purification and identification of RNA-binding proteins. (a) Examples of purification schemes for RNA-binding proteins using in vitro and in vivo approaches. For in vitro approaches, a tagged RNA construct is generated and bound to a solid support. In this example the MS2 protein-RNA interaction tagging method is shown with the target RNA (red), MS2-binding motif (purple) and MS2 protein (gray). Cell lysate is prepared and proteins from lysate are captured using the tagged RNA in vitro. For in vivo approaches, the target RNA is crosslinked to specific interacting RNA-binding proteins in living cells using UV, formaldehyde or other cross-linkers. Cells are lysed and the RNA-protein complexes captured from solution. In both scenarios, the complex is washed to remove non-specific interactions (green proteins). Finally the bound proteins are eluted. (b) MS is commonly used to identify the RBPs in a purified sample. In non-quantitative MS approaches, RBPs are purified from unlabeled cell material using either an RNA of interest or a control construct. After separation by one-dimensional gel electrophoresis, specific protein bands from the sample are selected, excised and identified by MS analysis. In quantitative MS approaches, proteins are differentially labeled based on their initial cell populations. Experimental and control purifications are performed on these labeled populations and the purified RBPs are pooled to create a single sample. MS analysis allows direct comparison of labeled peptides, which can then be quantified to determine specific proteins in the sample compared with the control. SILAC, stable isotope labeling by amino acids in cell culture.
References
-
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
