

The influence of CpG and UpA dinucleotide frequencies on RNA virus replication and characterization of the innate cellular pathways underlying virus attenuation and enhanced replication
- PMID: 24470146
- PMCID: PMC3985648
- DOI: 10.1093/nar/gku075
The influence of CpG and UpA dinucleotide frequencies on RNA virus replication and characterization of the innate cellular pathways underlying virus attenuation and enhanced replication
Abstract
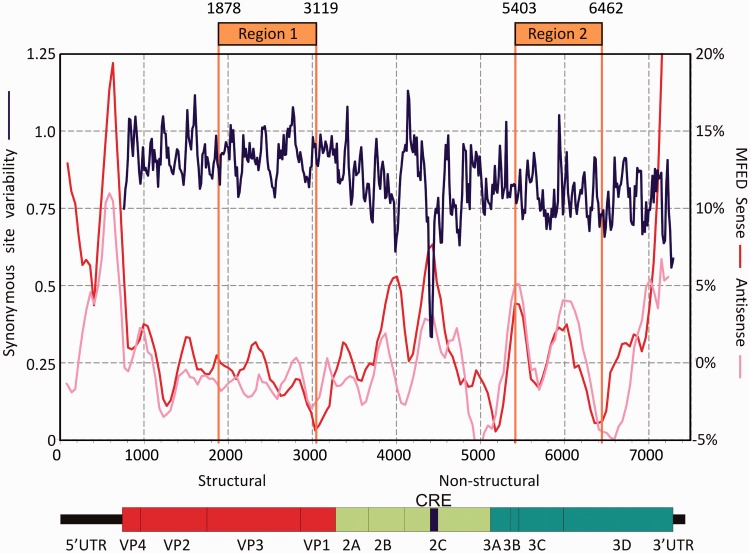
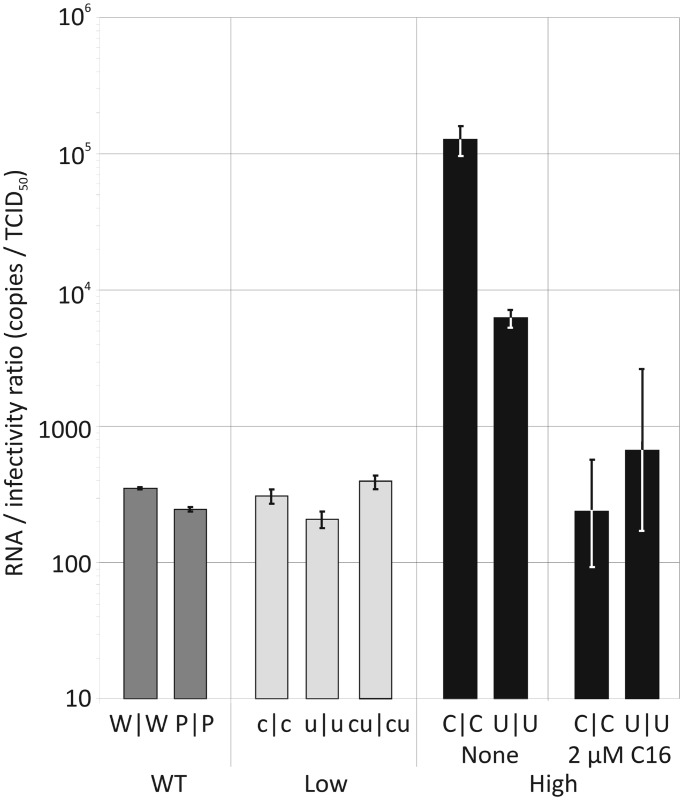
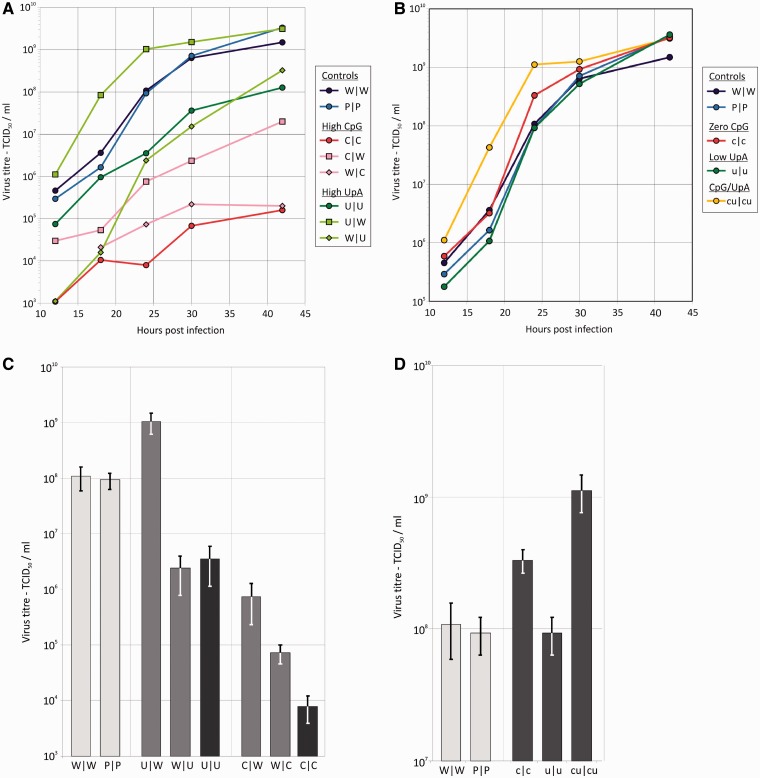
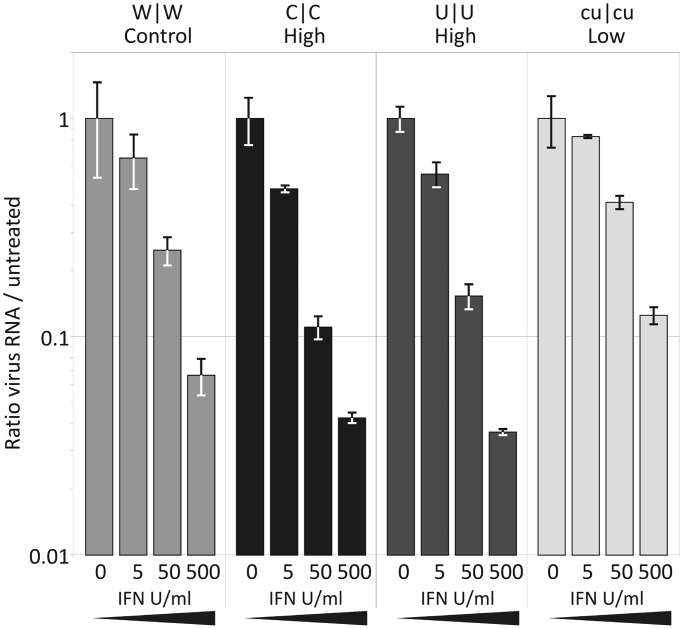
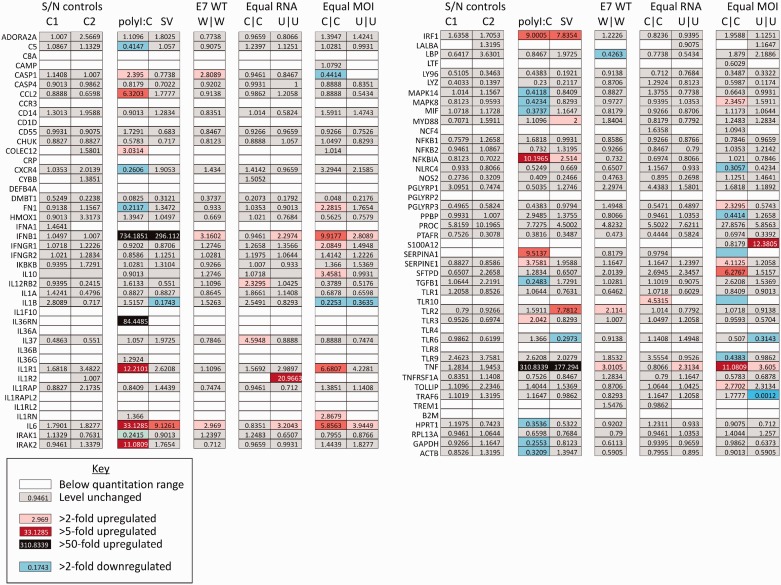
Most RNA viruses infecting mammals and other vertebrates show profound suppression of CpG and UpA dinucleotide frequencies. To investigate this functionally, mutants of the picornavirus, echovirus 7 (E7), were constructed with altered CpG and UpA compositions in two 1.1-1.3 Kbase regions. Those with increased frequencies of CpG and UpA showed impaired replication kinetics and higher RNA/infectivity ratios compared with wild-type virus. Remarkably, mutants with CpGs and UpAs removed showed enhanced replication, larger plaques and rapidly outcompeted wild-type virus on co-infections. Luciferase-expressing E7 sub-genomic replicons with CpGs and UpAs removed from the reporter gene showed 100-fold greater luminescence. E7 and mutants were equivalently sensitive to exogenously added interferon-β, showed no evidence for differential recognition by ADAR1 or pattern recognition receptors RIG-I, MDA5 or PKR. However, kinase inhibitors roscovitine and C16 partially or entirely reversed the attenuated phenotype of high CpG and UpA mutants, potentially through inhibition of currently uncharacterized pattern recognition receptors that respond to RNA composition. Generating viruses with enhanced replication kinetics has applications in vaccine production and reporter gene construction. More fundamentally, the findings introduce a new evolutionary paradigm where dinucleotide composition of viral genomes is subjected to selection pressures independently of coding capacity and profoundly influences host-pathogen interactions.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
