

Malignant peripheral nerve sheath tumors
- PMID: 24470531
- PMCID: PMC3926794
- DOI: 10.1634/theoncologist.2013-0328
Malignant peripheral nerve sheath tumors
Abstract
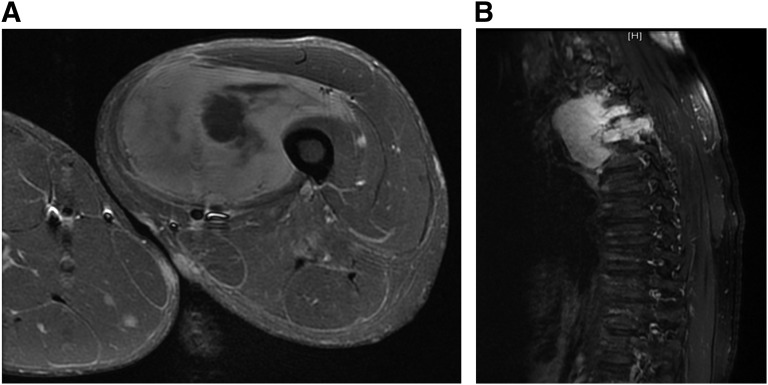
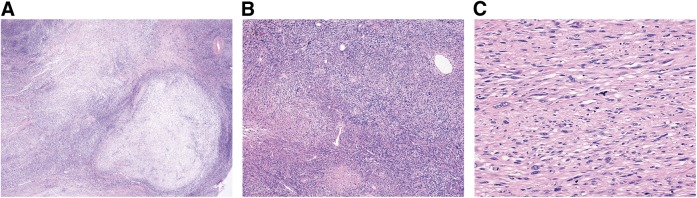
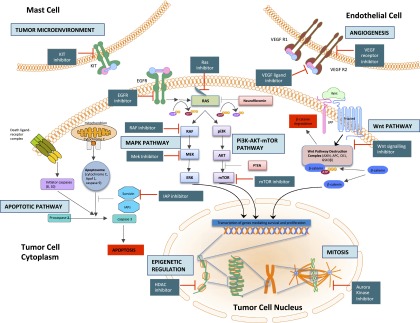
Malignant peripheral nerve sheath tumors (MPNST) are uncommon, biologically aggressive soft tissue sarcomas of neural origin that pose tremendous challenges to effective therapy. In 50% of cases, they occur in the context of neurofibromatosis type I, characterized by loss of function mutations to the tumor suppressor neurofibromin; the remainder arise sporadically or following radiation therapy. Prognosis is generally poor, with high rates of relapse following multimodality therapy in early disease, low response rates to cytotoxic chemotherapy in advanced disease, and propensity for rapid disease progression and high mortality. The last few years have seen an explosion in data surrounding the potential molecular drivers and targets for therapy above and beyond neurofibromin loss. These data span multiple nodes at various levels of cellular control, including major signal transduction pathways, angiogenesis, apoptosis, mitosis, and epigenetics. These include classical cancer-driving genetic aberrations such as TP53 and phosphatase and tensin homolog (PTEN) loss of function, and upregulation of mitogen-activated protein kinase (MAPK) and (mechanistic) target of rapamycin (TOR) pathways, as well as less ubiquitous molecular abnormalities involving inhibitors of apoptosis proteins, aurora kinases, and the Wingless/int (Wnt) signaling pathway. We review the current understanding of MPNST biology, current best practices of management, and recent research developments in this disease, with a view to informing future advancements in patient care.
Keywords: Clinical trials; Malignant peripheral nerve sheath tumor; Molecular targeted therapy; Neurofibromatosis type 1; Sarcoma.
Conflict of interest statement
Disclosures of potential conflicts of interest may be found at the end of this article.
Figures
Similar articles
-
Systemic Options for Malignant Peripheral Nerve Sheath Tumors.Curr Treat Options Oncol. 2021 Feb 27;22(4):33. doi: 10.1007/s11864-021-00830-7. Curr Treat Options Oncol. 2021. PMID: 33641042 Review.
-
[Targeted therapy for malignant peripheral nerve sheath tumor: translational research and clinical application].Zhonghua Zhong Liu Za Zhi. 2019 Sep 23;41(9):648-653. doi: 10.3760/cma.j.issn.0253-3766.2019.09.002. Zhonghua Zhong Liu Za Zhi. 2019. PMID: 31550853 Review. Chinese.
-
Malignant peripheral nerve sheath tumour (MPNST): the clinical implications of cellular signalling pathways.Expert Rev Mol Med. 2009 Oct 19;11:e30. doi: 10.1017/S1462399409001227. Expert Rev Mol Med. 2009. PMID: 19835664 Review.
-
Malignant peripheral nerve sheath tumor: models, biology, and translation.Oncogene. 2022 Apr;41(17):2405-2421. doi: 10.1038/s41388-022-02290-1. Epub 2022 Apr 7. Oncogene. 2022. PMID: 35393544 Free PMC article. Review.
-
Clinical genomic profiling identifies TYK2 mutation and overexpression in patients with neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors.Cancer. 2017 Apr 1;123(7):1194-1201. doi: 10.1002/cncr.30455. Epub 2016 Nov 22. Cancer. 2017. PMID: 27875628 Free PMC article.
Cited by
-
Denosumab combined with chemotherapy followed by anlotinib in the treatment of multiple metastases of malignant peripheral nerve sheath tumor: a case report and literature review.Front Oncol. 2024 Jul 25;14:1399021. doi: 10.3389/fonc.2024.1399021. eCollection 2024. Front Oncol. 2024. PMID: 39119091 Free PMC article.
-
A rare case of cecal malignant peripheral nerve sheath tumor in a dog.J Vet Med Sci. 2022 Aug 1;84(8):1051-1055. doi: 10.1292/jvms.22-0042. Epub 2022 Jun 21. J Vet Med Sci. 2022. PMID: 35732442 Free PMC article.
-
Two case reports of fair lower limb function after sciatic nerve sacrifice for the treatment of a malignant peripheral nerve sheath tumor.Int Cancer Conf J. 2018 Jul 13;7(4):137-141. doi: 10.1007/s13691-018-0338-x. eCollection 2018 Oct. Int Cancer Conf J. 2018. PMID: 31149533 Free PMC article.
-
Imaging of Peripheral Intraneural Tumors: A Comprehensive Review for Radiologists.Cancers (Basel). 2025 Jan 13;17(2):246. doi: 10.3390/cancers17020246. Cancers (Basel). 2025. PMID: 39858028 Free PMC article. Review.
-
Close Follow-Up of Patients with Neurofibromatosis Type 1 Reduces the Incidence of Malignant Peripheral Nerve Sheath Tumour.Cancers (Basel). 2025 Apr 12;17(8):1306. doi: 10.3390/cancers17081306. Cancers (Basel). 2025. PMID: 40282482 Free PMC article.
References
-
- Fletcher CDM, Bridge JA, Hogendoorn PCW. et al. WHO Classification of Tumours of Soft Tissue and Bone. Lyon, France: IARC; 2013.
-
- Ng VY, Scharschmidt TJ, Mayerson JL, et al. Incidence and survival in sarcoma in the United States: A focus on musculoskeletal lesions. Anticancer Res. 2013;33:2597–2604. - PubMed
-
- Cichowski K, Shih TS, Schmitt E, et al. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–2176. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
