

Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms
- PMID: 24470592
- PMCID: PMC3968390
- DOI: 10.1182/blood-2014-01-547760
Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms
Abstract
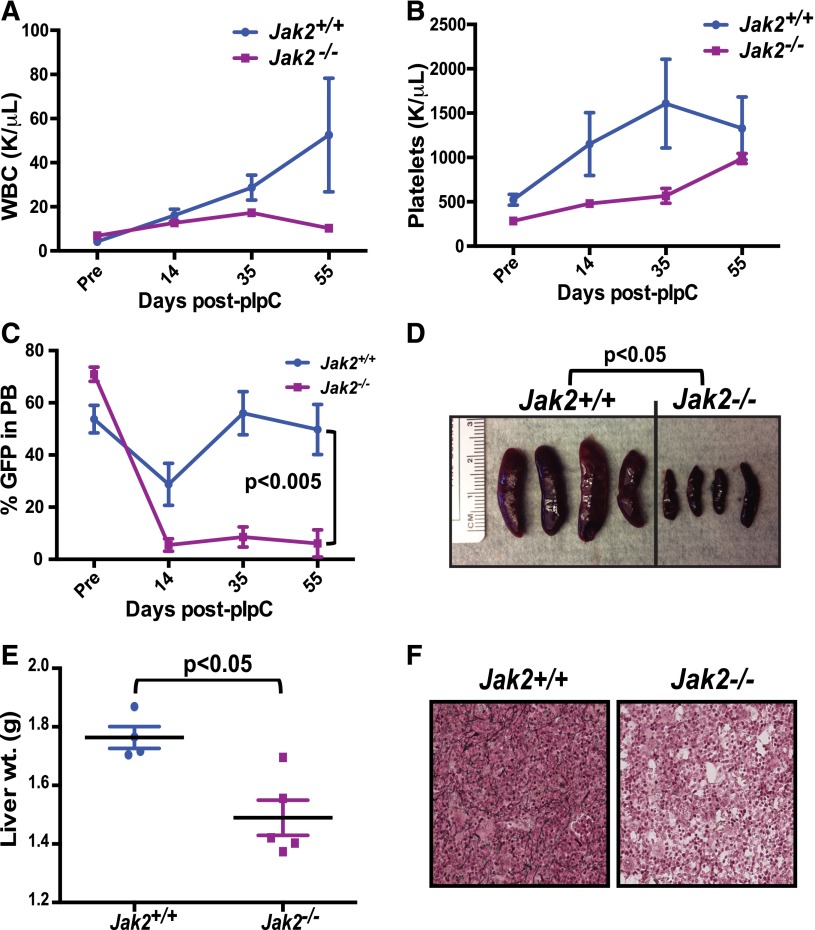
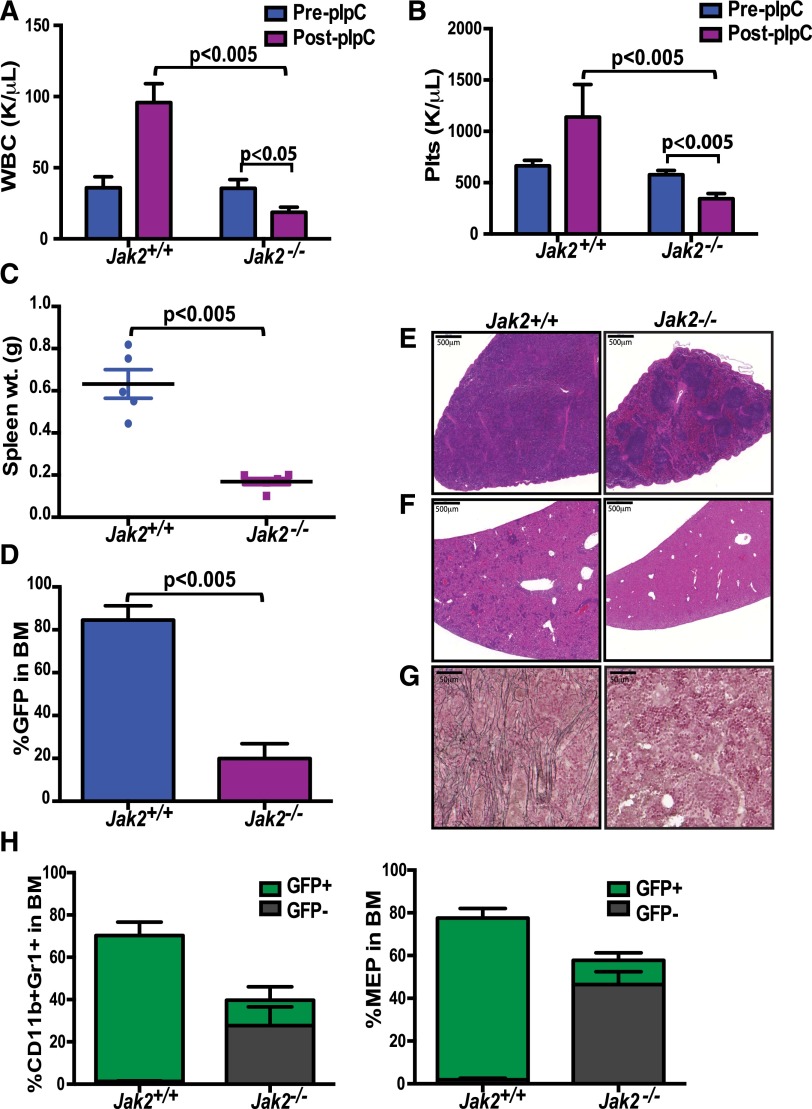
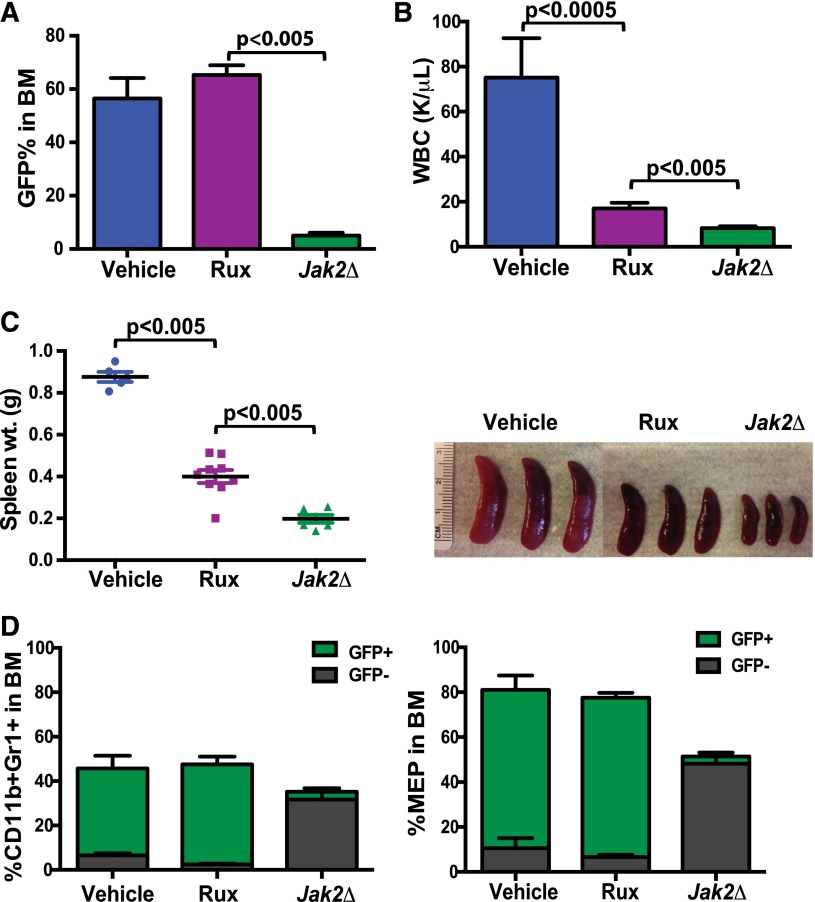
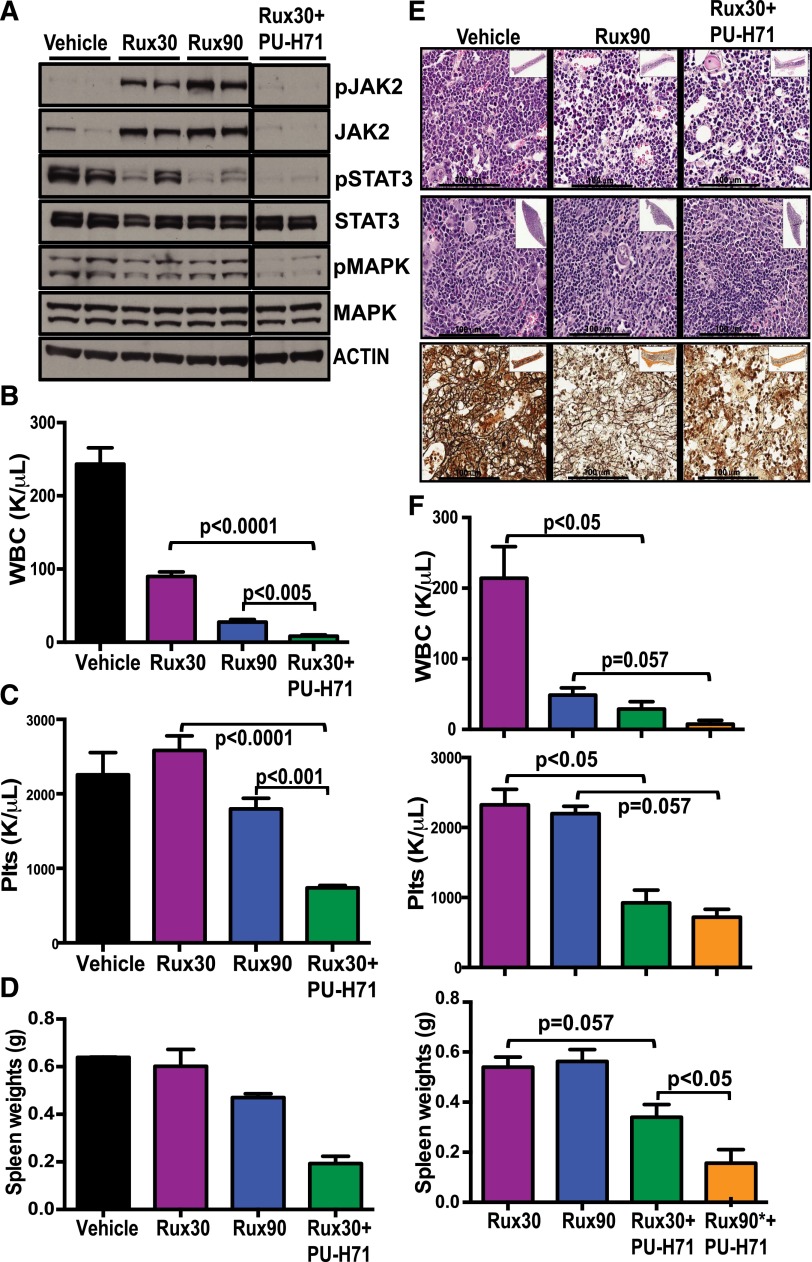
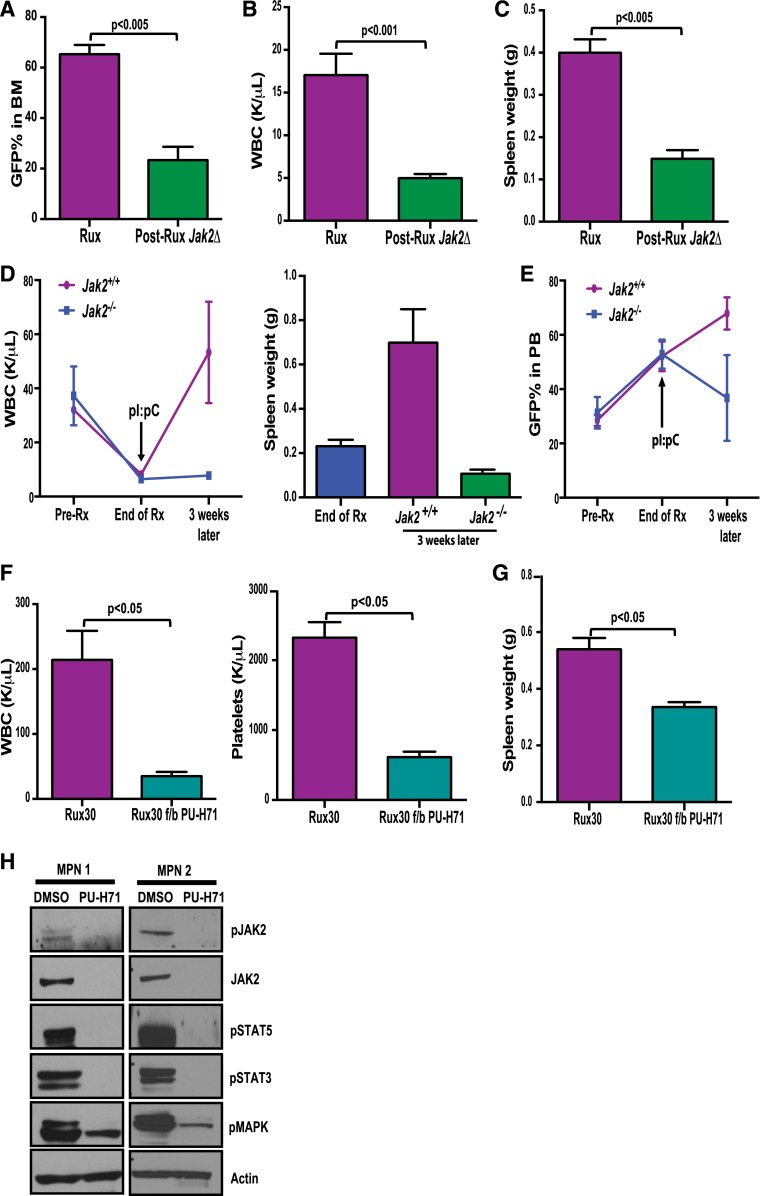
The discovery of JAK2/MPL mutations in patients with myeloproliferative neoplasms (MPN) led to clinical development of Janus kinase (JAK) inhibitors for treatment of MPN. These inhibitors improve constitutional symptoms and splenomegaly but do not significantly reduce mutant allele burden in patients. We recently showed that chronic exposure to JAK inhibitors results in inhibitor persistence via JAK2 transactivation and persistent JAK-signal transducer and activator of transcription signaling. We performed genetic and pharmacologic studies to determine whether improved JAK2 inhibition would show increased efficacy in MPN models and primary samples. Jak2 deletion in vivo led to profound reduction in disease burden not seen with JAK inhibitors, and deletion of Jak2 following chronic ruxolitinib therapy markedly reduced mutant allele burden. This demonstrates that JAK2 remains an essential target in MPN cells that survive in the setting of chronic JAK inhibition. Combination therapy with the heat shock protein 90 (HSP90) inhibitor PU-H71 and ruxolitinib reduced total and phospho-JAK2 and achieved more potent inhibition of downstream signaling than ruxolitinib monotherapy. Combination treatment improved blood counts, spleen weights, and reduced bone marrow fibrosis compared with ruxolitinib alone. These data suggest alternate approaches that increase JAK2 targeting, including combination JAK/HSP90 inhibitor therapy, are warranted in the clinical setting.
Figures
Comment in
-
Increasing therapeutic efficacy in MPN.Blood. 2014 Mar 27;123(13):1982-3. doi: 10.1182/blood-2014-02-554766. Blood. 2014. PMID: 24677402
References
-
- Baxter EJ, Scott LM, Campbell PJ, et al. Cancer Genome Project. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. - PubMed
-
- James C, Ugo V, Le Couédic J-P, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. - PubMed
-
- Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. - PubMed
-
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
