

The intra-host evolutionary and population dynamics of human immunodeficiency virus type 1: a phylogenetic perspective
- PMID: 24470967
- PMCID: PMC3892624
- DOI: 10.4081/idr.2013.s1.e3
The intra-host evolutionary and population dynamics of human immunodeficiency virus type 1: a phylogenetic perspective
Abstract
The intra-host evolutionary and population dynamics of the human immunodeficiency virus type 1 (HIV-1), the cause of the acquired immunodeficiency syndrome, have been the focus of one of the most extensive study efforts in the field of molecular evolution over the past three decades. As HIV-1 is among the fastest mutating organisms known, viral sequence data sampled over time from infected patients can provide, through phylogenetic analysis, significant insights about the tempo and mode of evolutionary processes shaped by complex interaction with the host milieu. Five main aspects are discussed: the patterns of HIV-1 intra-host diversity and divergence over time in relation to different phases of disease progression; the impact of selection on the temporal structure of HIV-1 intra-host genealogies inferred from longitudinally sampled viral sequences; HIV-1 intra-host sub-population structure; the potential relationship between viral evolutionary rate and disease progression and the central evolutionary role played by recombination occurring in super-infected cells.
Keywords: HIV-1; intrahost evolution; phylogenetic analysis; population dynamics.
Figures
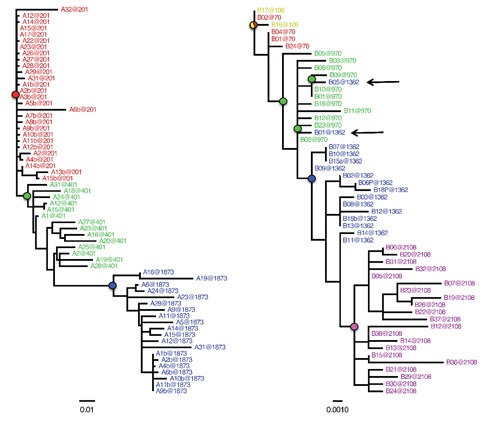
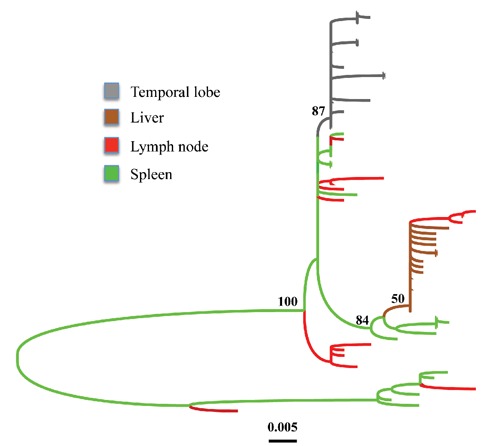
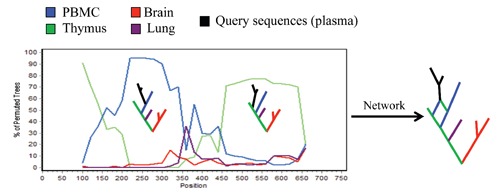
References
-
- Barré-Sinoussi F, Chermann JC, Rey F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for the acquired immunodeficiency syndrome (AIDS). Science 1983;292:1102-05 - PubMed
-
- Hahn BH, Shaw GM, Taylor ME, et al. Genetic variation in HTLV-III/LAV over time in patients with AIDS or at risk for AIDS. Science 1986;232:1548-53 - PubMed
-
- Korber BT, Allen EE, Farmer AD, Myers GL. Heterogeneity of HIV-1 and HIV-2. AIDS 1995;9:S5-S18 - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous