

Angiotensin II type 1 and 2 receptors and lymphatic vessels modulate lung remodeling and fibrosis in systemic sclerosis and idiopathic pulmonary fibrosis
- PMID: 24473559
- PMCID: PMC3870314
- DOI: 10.6061/clinics/2014(01)07
Angiotensin II type 1 and 2 receptors and lymphatic vessels modulate lung remodeling and fibrosis in systemic sclerosis and idiopathic pulmonary fibrosis
Abstract
Objective: To validate the importance of the angiotensin II receptor isotypes and the lymphatic vessels in systemic sclerosis and idiopathic pulmonary fibrosis.
Methods: We examined angiotensin II type 1 and 2 receptors and lymphatic vessels in the pulmonary tissues obtained from open lung biopsies of 30 patients with systemic sclerosis and 28 patients with idiopathic pulmonary fibrosis. Their histologic patterns included cellular and fibrotic non-specific interstitial pneumonia for systemic sclerosis and usual interstitial pneumonia for idiopathic pulmonary fibrosis. We used immunohistochemistry and histomorphometry to evaluate the number of cells in the alveolar septae and the vessels stained by these markers. Survival curves were also used.
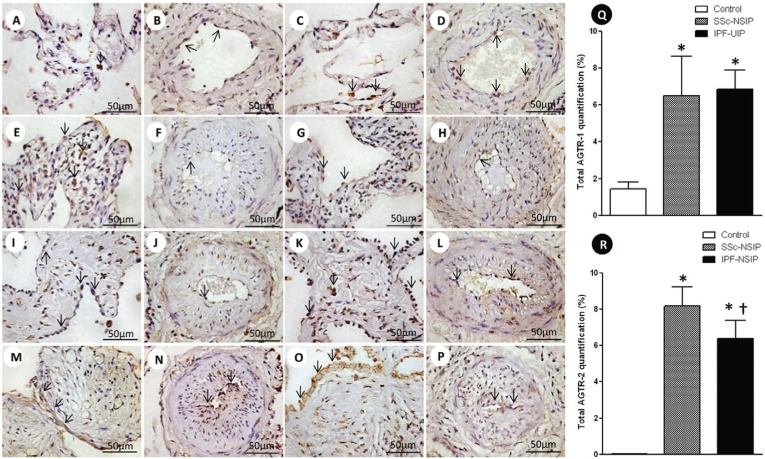
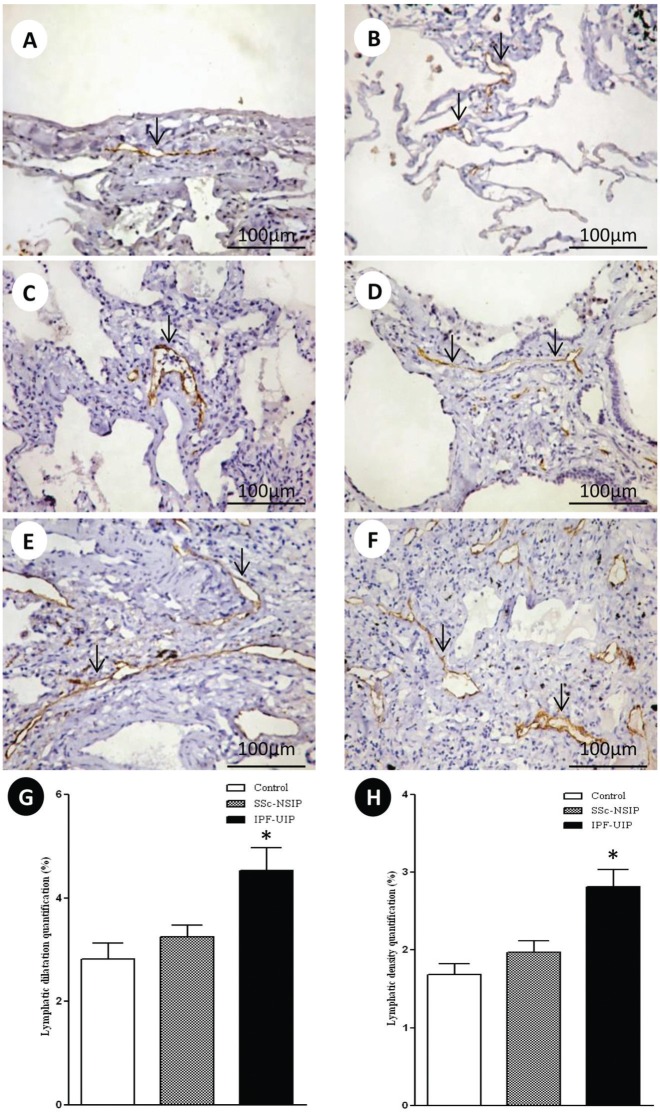
Results: We found a significantly increased percentage of septal and vessel cells immunostained for the angiotensin type 1 and 2 receptors in the systemic sclerosis and idiopathic pulmonary fibrosis patients compared with the controls. A similar percentage of angiotensin 2 receptor positive vessel cells was observed in fibrotic non-specific interstitial pneumonia and usual interstitial pneumonia. A significantly increased percentage of lymphatic vessels was present in the usual interstitial pneumonia group compared with the non-specific interstitial pneumonia and control groups. A Cox regression analysis showed a high risk of death for the patients with usual interstitial pneumonia and a high percentage of vessel cells immunostained for the angiotensin 2 receptor in the lymphatic vessels.
Conclusion: We concluded that angiotensin II receptor expression in the lung parenchyma can potentially control organ remodeling and fibrosis, which suggests that strategies aimed at preventing high angiotensin 2 receptor expression may be used as potential therapeutic target in patients with pulmonary systemic sclerosis and idiopathic pulmonary fibrosis.
Conflict of interest statement
No potential conflict of interest was reported.
Figures
References
-
- American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165(2):277–304. - PubMed
-
- Douglas WW, Tazelaar HD, Hartman TE, Hartman RP, Decker PA, Schroeder DR, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med. 2001;164(7):1182–5. - PubMed
-
- Flaherty KR, Colby TV, Travis WD, Toews GB, Mumford J, Murray S, et al. Fibroblastic foci in usual interstitial pneumonia: idiopathic versus collagen vascular disease. Am J Respir Crit Care Med. 2003;167(10):1410–5. - PubMed
-
- Papiris SA, Vlachoyiannopoulos PG, Maniati MA, Karakostas KX, Constantopoulos SH, Moutsopoulos HH. Idiopathic pulmonary fibrosis and pulmonary fibrosis in diffuse systemic sclerosis: two fibroses with different prognoses. Respiration. 1997;64(1):81–5. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
