

Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline
- PMID: 24475911
- PMCID: PMC3922167
- DOI: 10.1186/1471-2105-15-30
Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline
Abstract
Background: Massively parallel DNA sequencing generates staggering amounts of data. Decreasing cost, increasing throughput, and improved annotation have expanded the diversity of genomics applications in research and clinical practice. This expanding scale creates analytical challenges: accommodating peak compute demand, coordinating secure access for multiple analysts, and sharing validated tools and results.
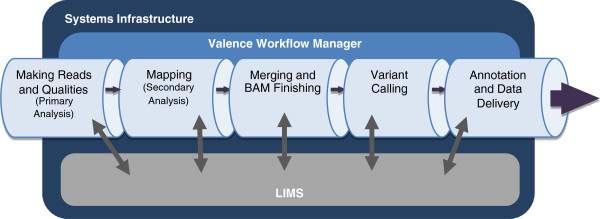
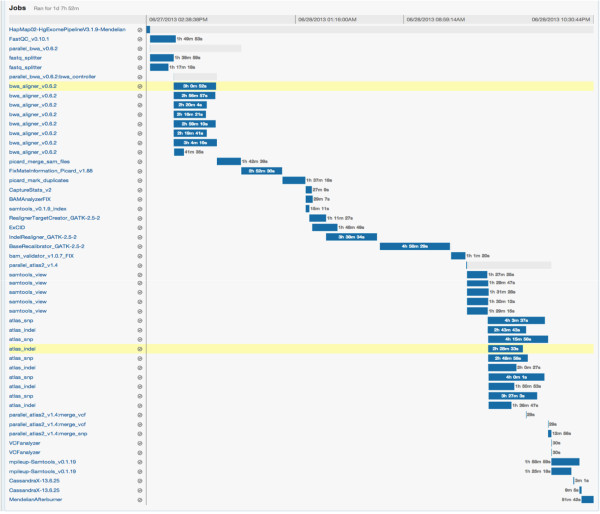
Results: To address these challenges, we have developed the Mercury analysis pipeline and deployed it in local hardware and the Amazon Web Services cloud via the DNAnexus platform. Mercury is an automated, flexible, and extensible analysis workflow that provides accurate and reproducible genomic results at scales ranging from individuals to large cohorts.
Conclusions: By taking advantage of cloud computing and with Mercury implemented on the DNAnexus platform, we have demonstrated a powerful combination of a robust and fully validated software pipeline and a scalable computational resource that, to date, we have applied to more than 10,000 whole genome and whole exome samples.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
