

Landscape and variation of RNA secondary structure across the human transcriptome
- PMID: 24476892
- PMCID: PMC3973747
- DOI: 10.1038/nature12946
Landscape and variation of RNA secondary structure across the human transcriptome
Abstract
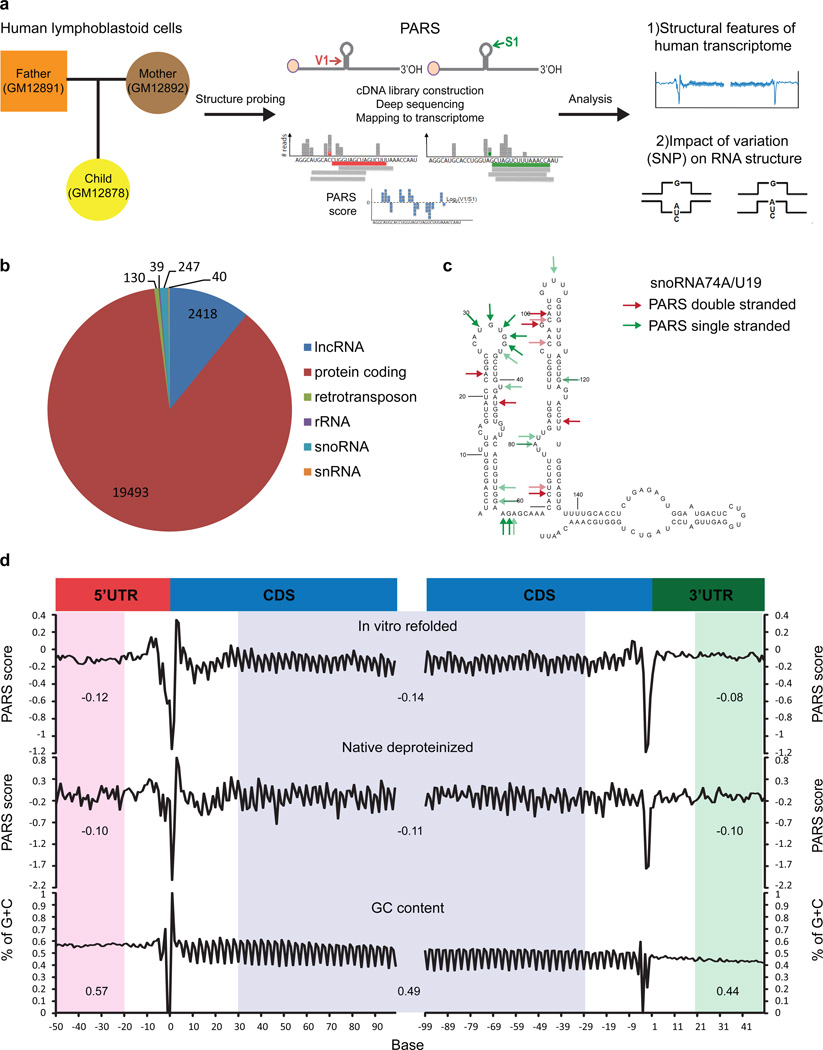
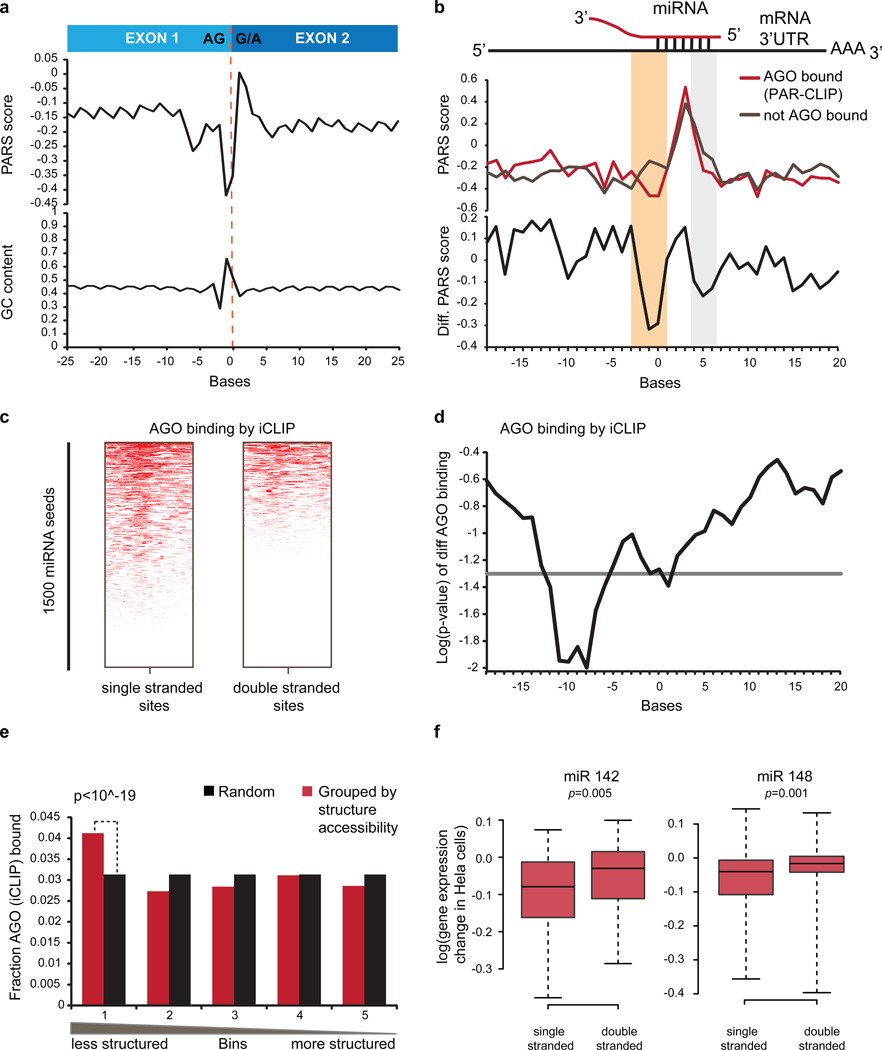
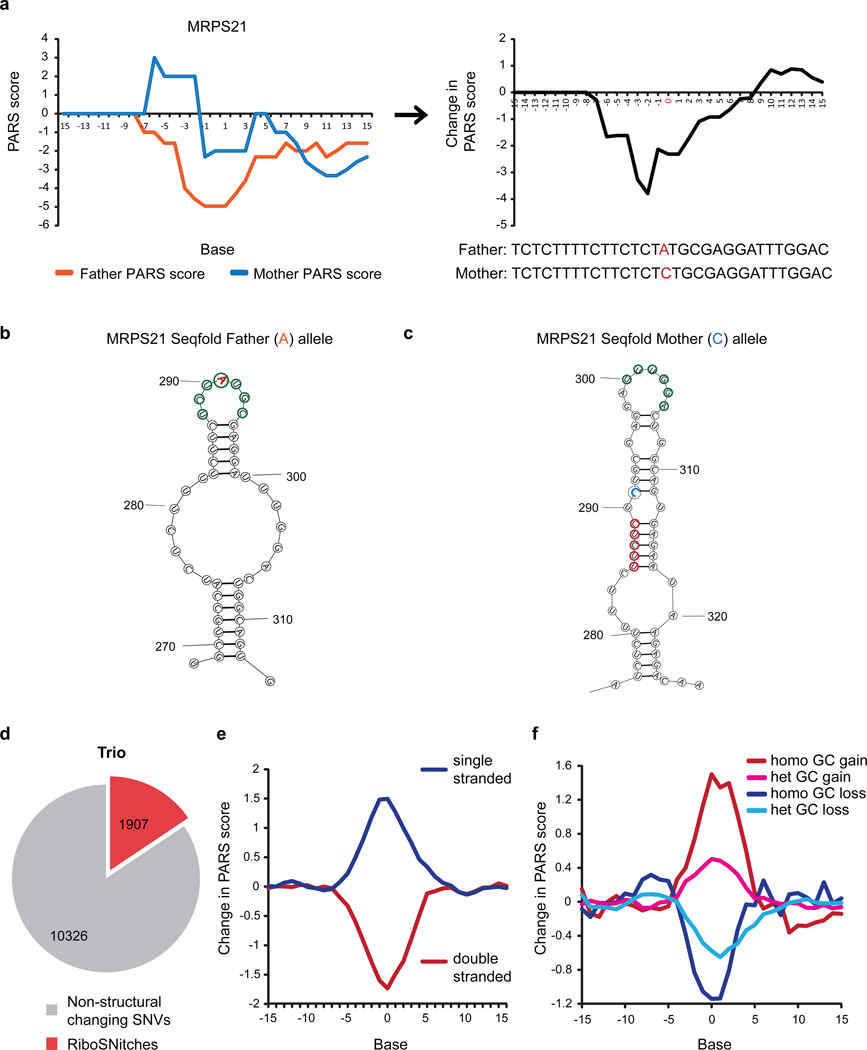
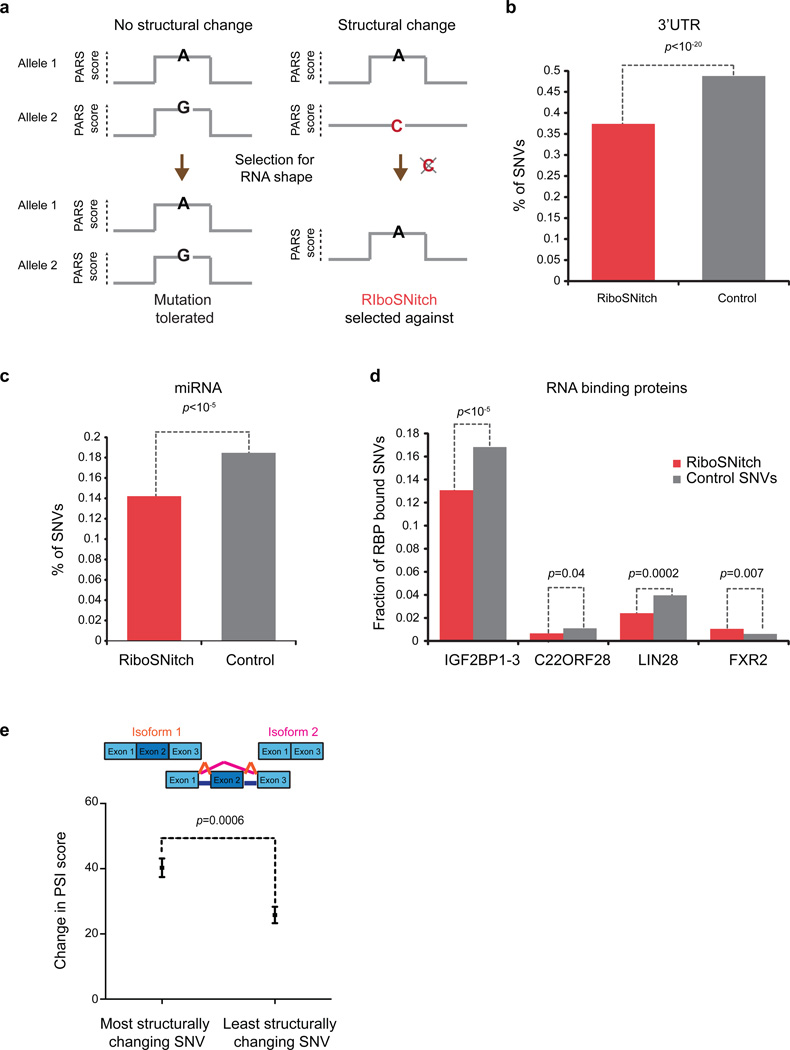
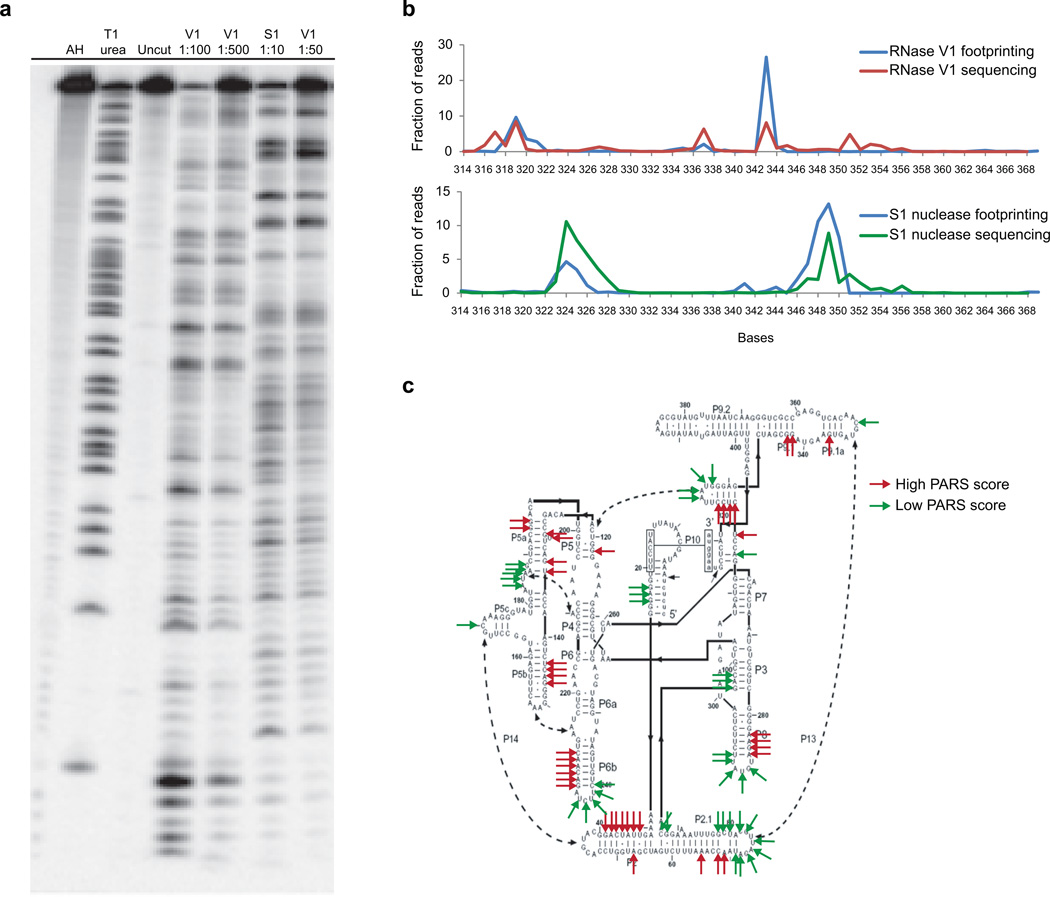
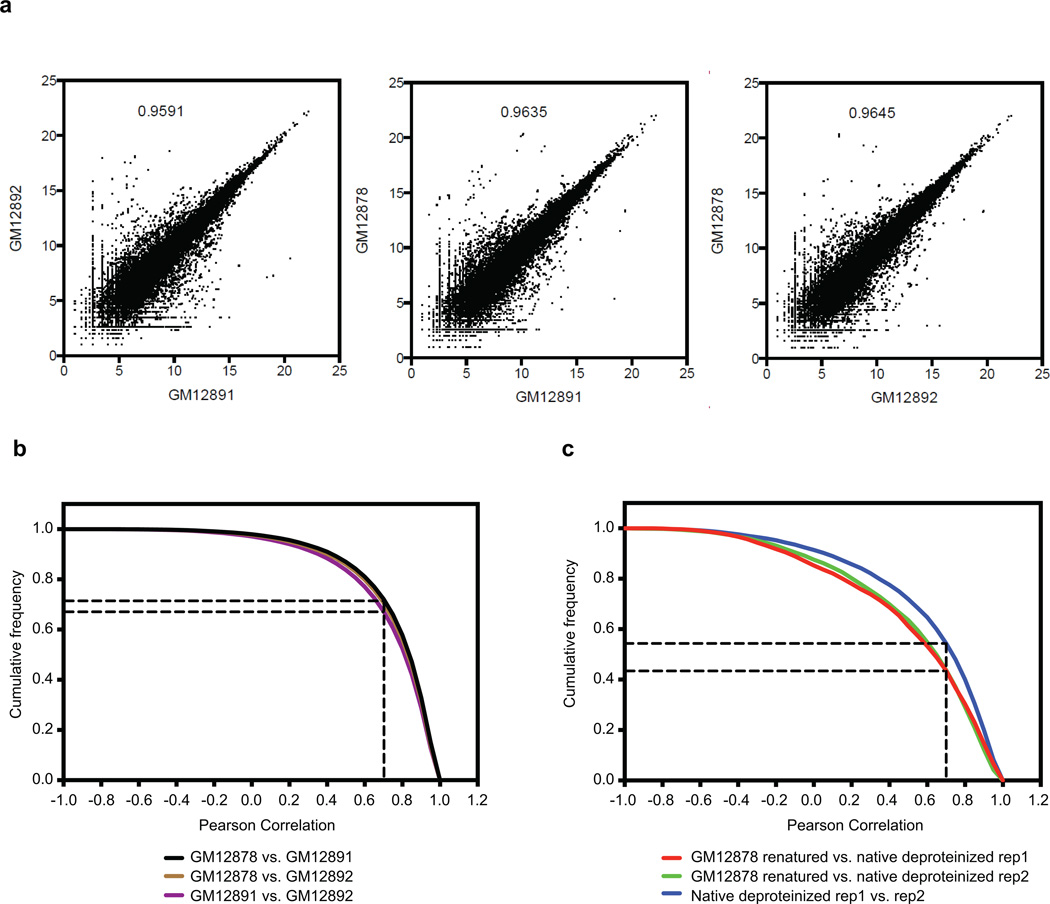
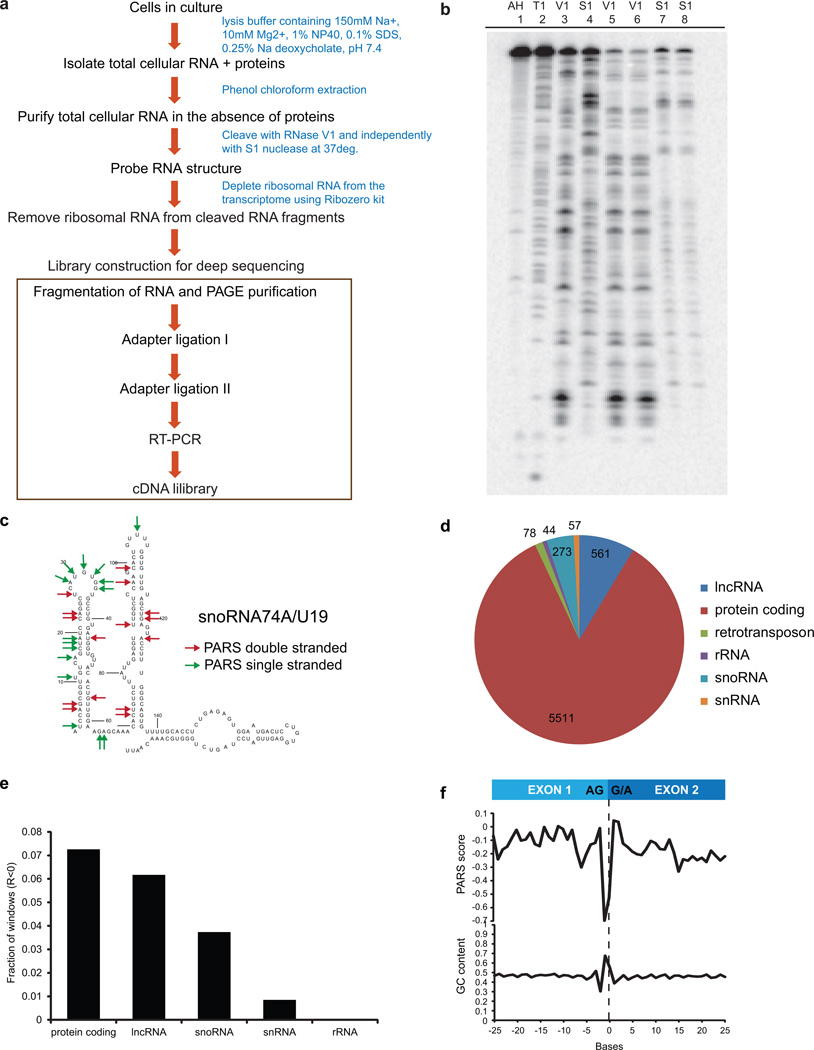
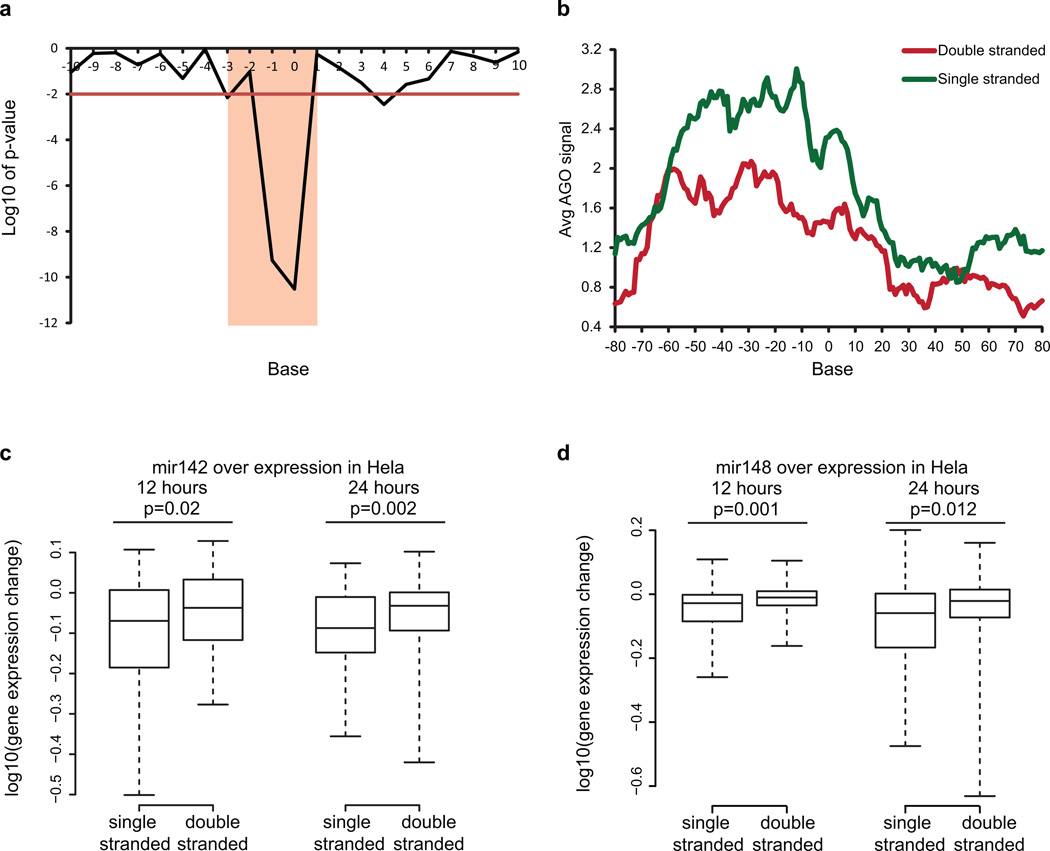
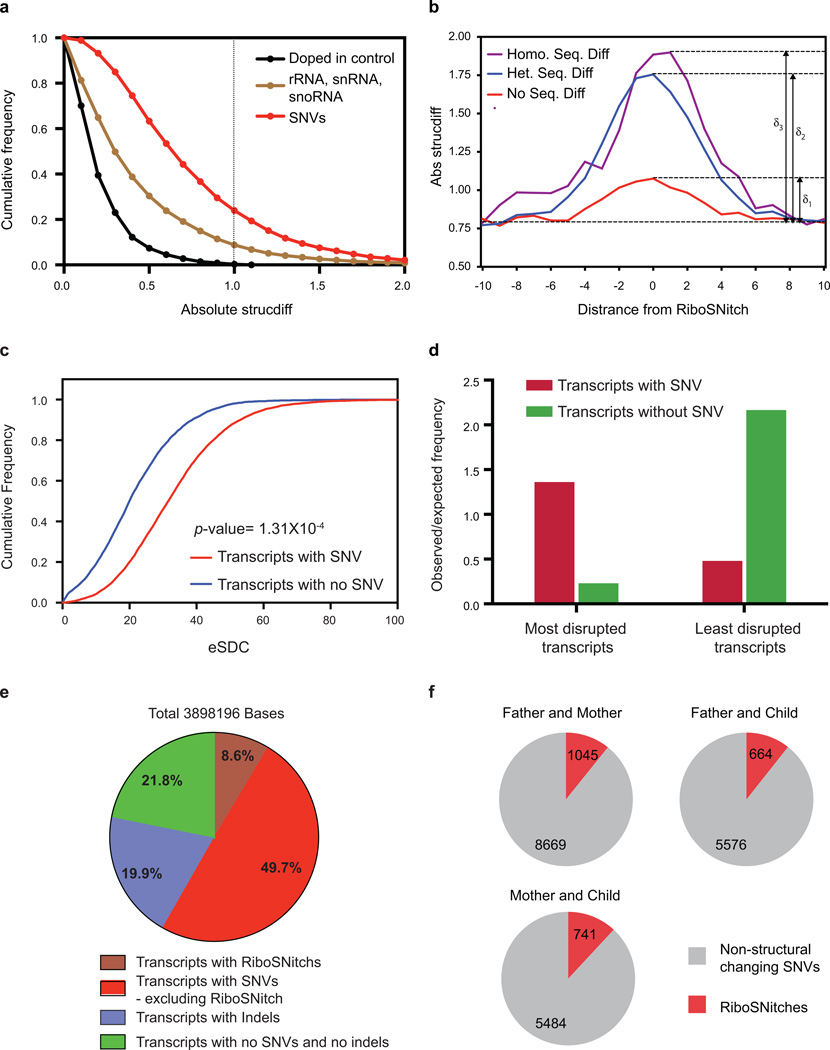
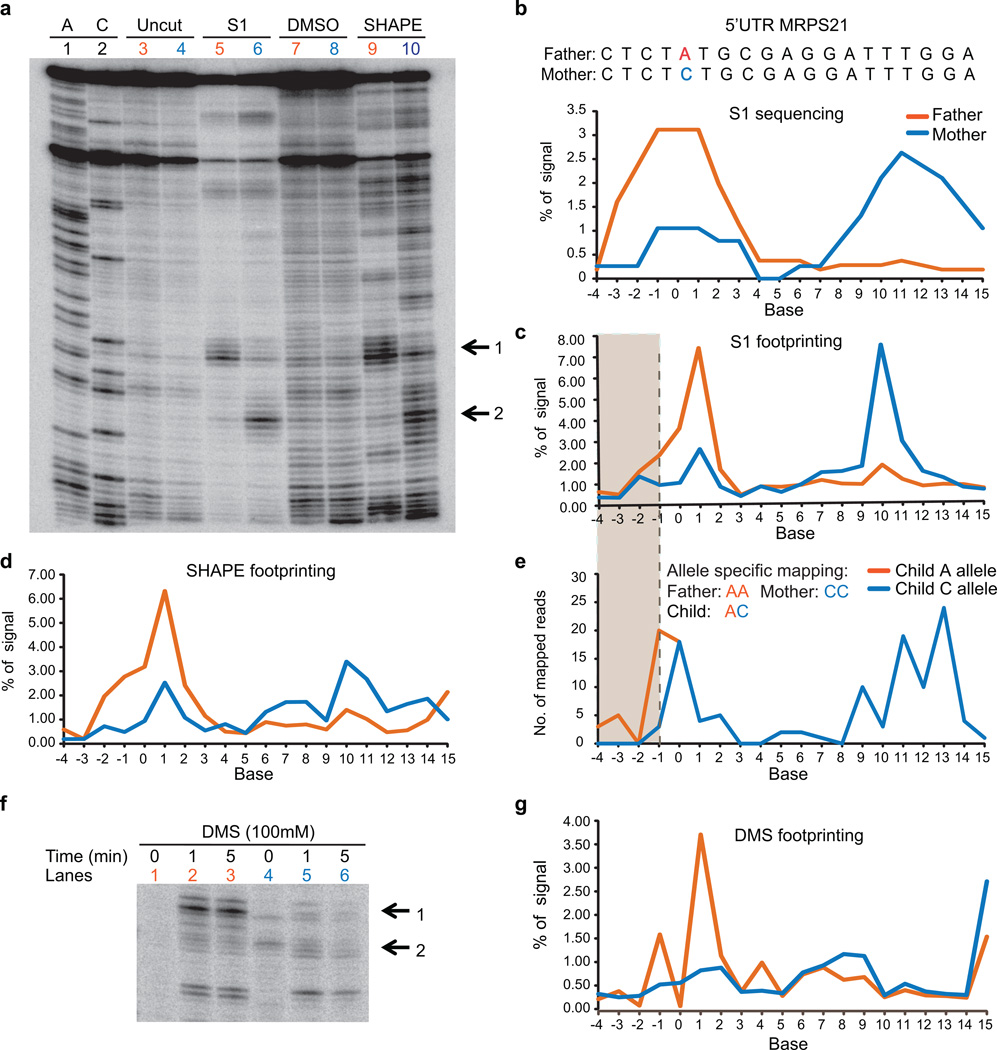
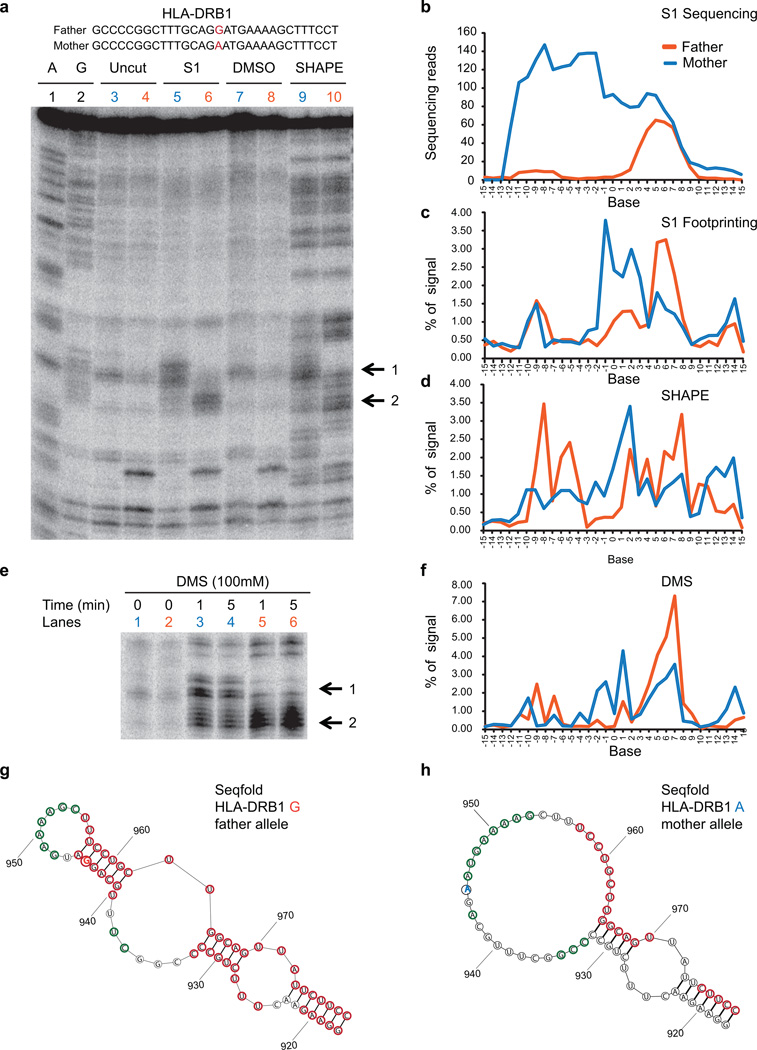
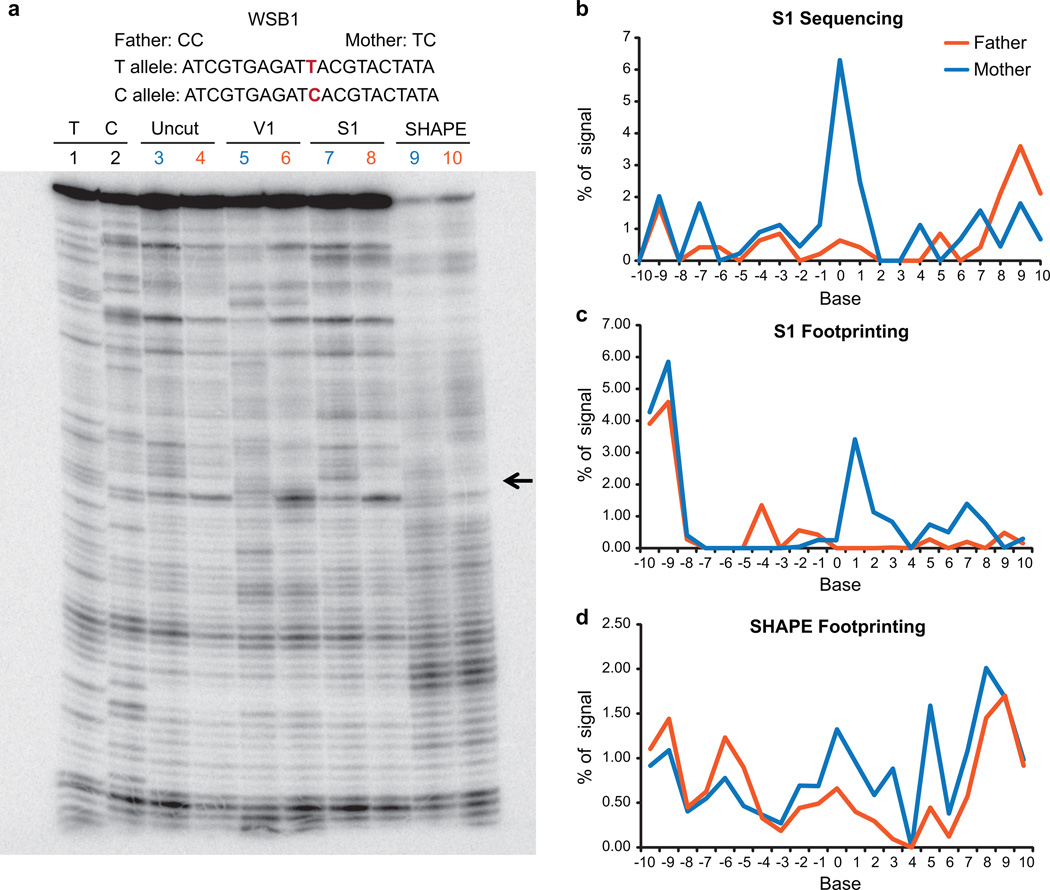
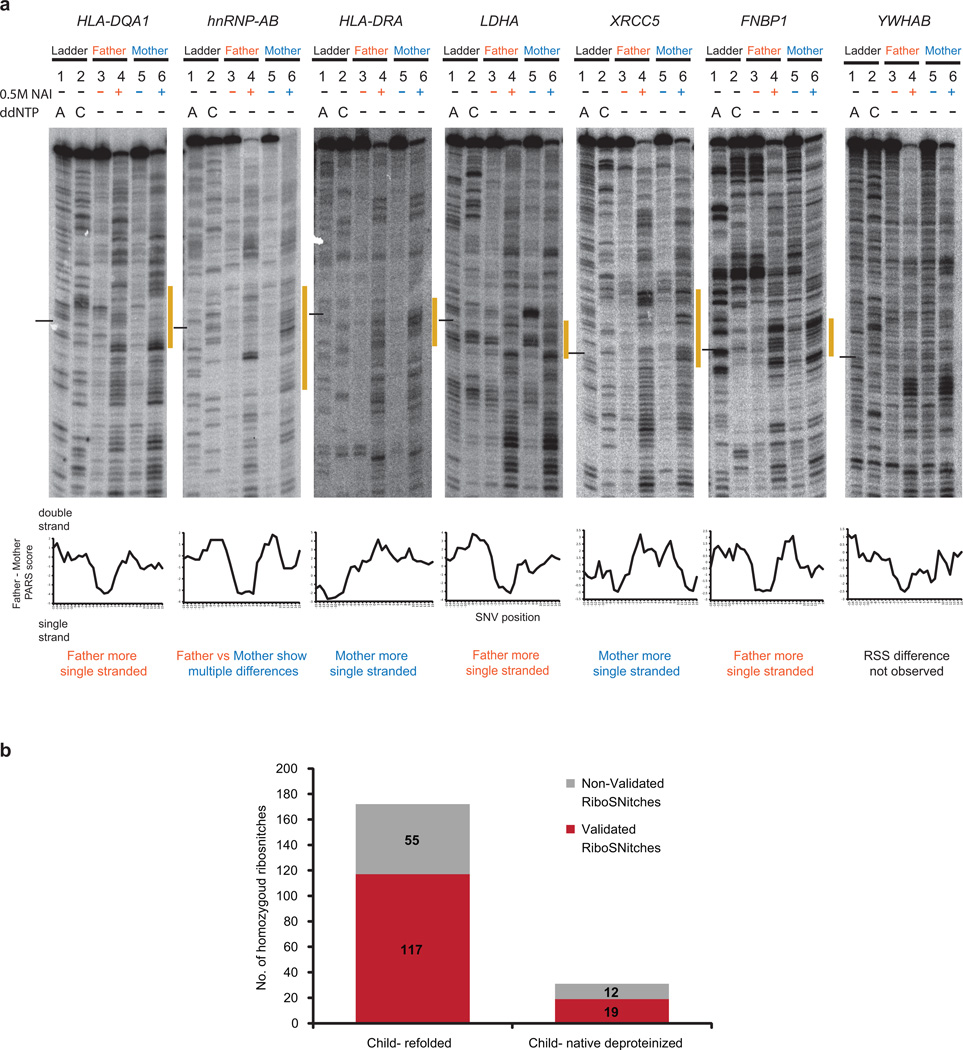
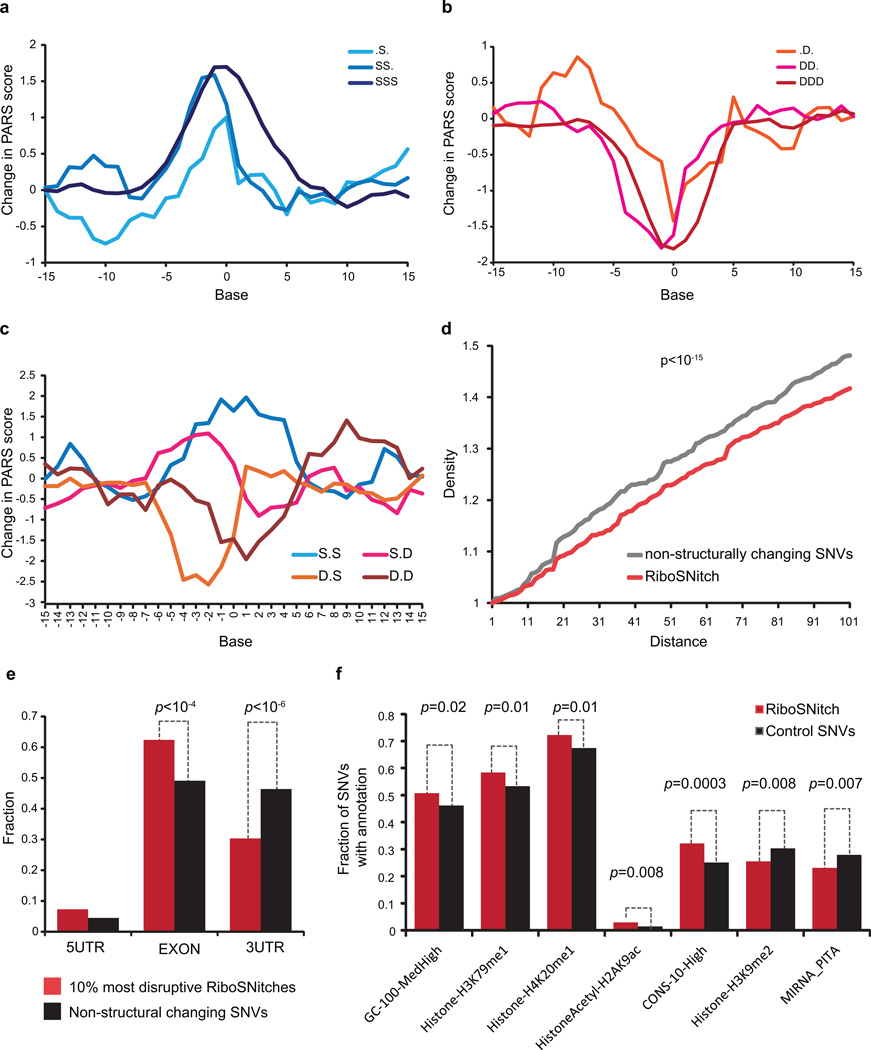
In parallel to the genetic code for protein synthesis, a second layer of information is embedded in all RNA transcripts in the form of RNA structure. RNA structure influences practically every step in the gene expression program. However, the nature of most RNA structures or effects of sequence variation on structure are not known. Here we report the initial landscape and variation of RNA secondary structures (RSSs) in a human family trio (mother, father and their child). This provides a comprehensive RSS map of human coding and non-coding RNAs. We identify unique RSS signatures that demarcate open reading frames and splicing junctions, and define authentic microRNA-binding sites. Comparison of native deproteinized RNA isolated from cells versus refolded purified RNA suggests that the majority of the RSS information is encoded within RNA sequence. Over 1,900 transcribed single nucleotide variants (approximately 15% of all transcribed single nucleotide variants) alter local RNA structure. We discover simple sequence and spacing rules that determine the ability of point mutations to impact RSSs. Selective depletion of 'riboSNitches' versus structurally synonymous variants at precise locations suggests selection for specific RNA shapes at thousands of sites, including 3' untranslated regions, binding sites of microRNAs and RNA-binding proteins genome-wide. These results highlight the potentially broad contribution of RNA structure and its variation to gene regulation.
Figures
Comment in
-
Molecular biology: A second layer of information in RNA.Nature. 2014 Jan 30;505(7485):621-2. doi: 10.1038/505621a. Nature. 2014. PMID: 24476882 Free PMC article.
-
RNA: riboSNitches reveal heredity in RNA secondary structure.Nat Rev Genet. 2014 Apr;15(4):219. doi: 10.1038/nrg3700. Epub 2014 Feb 18. Nat Rev Genet. 2014. PMID: 24535248 No abstract available.
References
References for full methods
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
