

Factors associated with survival in a contemporary adult sickle cell disease cohort
- PMID: 24478166
- PMCID: PMC3988218
- DOI: 10.1002/ajh.23683
Factors associated with survival in a contemporary adult sickle cell disease cohort
Abstract
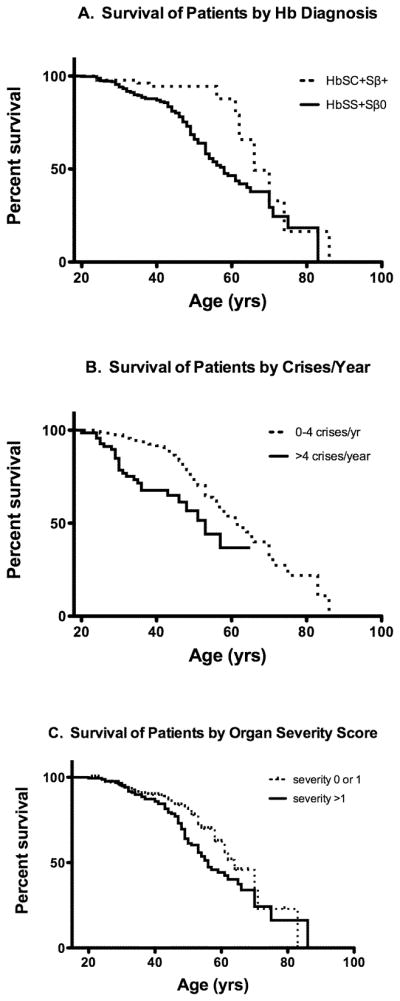
In this study, the relationship of clinical differences among patients with sickle cell disease (SCD) was examined to understand the major contributors to early mortality in a contemporary cohort. Survival data were obtained for 542 adult subjects who were enrolled since 2002 at three university hospitals in the southeast United States. Subjects were followed up for a median of 9.3 years. At enrollment, clinical parameters were collected, including hemoglobin (Hb) genotype, baseline laboratory values, comorbidities, and medication usage. Levels of soluble adhesion molecules were measured for a subset of 87 subjects. The relationship of clinical characteristics to survival was determined using regression analysis. Median age at enrollment was 32 years. Median survival was 61 years for all subjects. Median survival for Hb SS and Sβ(0) was 58 years and for Hb SC and Sβ(+) was 66 years. Elevated white blood count, lower estimated glomerular filtration rate, proteinuria, frequency of pain crises, pulmonary hypertension, cerebrovascular events, seizures, stroke, sVCAM-1, and short-acting narcotics use were significantly associated with decreased survival. Forty-two percent of subjects were on hydroxyurea therapy, which was not associated with survival. SCD continues to reduce life expectancy for affected individuals, particularly those with Hb Sβ(0) and SS. Not only were comorbidities individually associated with decreased survival but also an additive effect was observed, thus, those with a greater number of negative endpoints had worse survival (P < 0.0001). The association of higher sVCAM-1 levels with decreased survival suggests that targeted therapies to reduce endothelial damage and inflammation may also be beneficial.
Copyright © 2014 Wiley Periodicals, Inc.
Figures
References
-
- Diggs L. Sickle Cell Disease: Diagnosis, Management, Education and Research. Mosby; 1973. pp. 189–229.
-
- Leikin SL, Gallagher D, Kinney TR, et al. Mortality in children and adolescents with sickle cell disease. Cooperative study of sickle cell disease. Pediatrics. 1989;84:500–508. - PubMed
-
- From the Centers for Disease Control and Prevention. Mortality among children with sickle cell disease identified by newborn screening during 1990–1994: California, Illinois, and New York. JAMA. 1998;279:1059–1060. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases