

A comparative study of Whi5 and retinoblastoma proteins: from sequence and structure analysis to intracellular networks
- PMID: 24478706
- PMCID: PMC3897220
- DOI: 10.3389/fphys.2013.00315
A comparative study of Whi5 and retinoblastoma proteins: from sequence and structure analysis to intracellular networks
Abstract
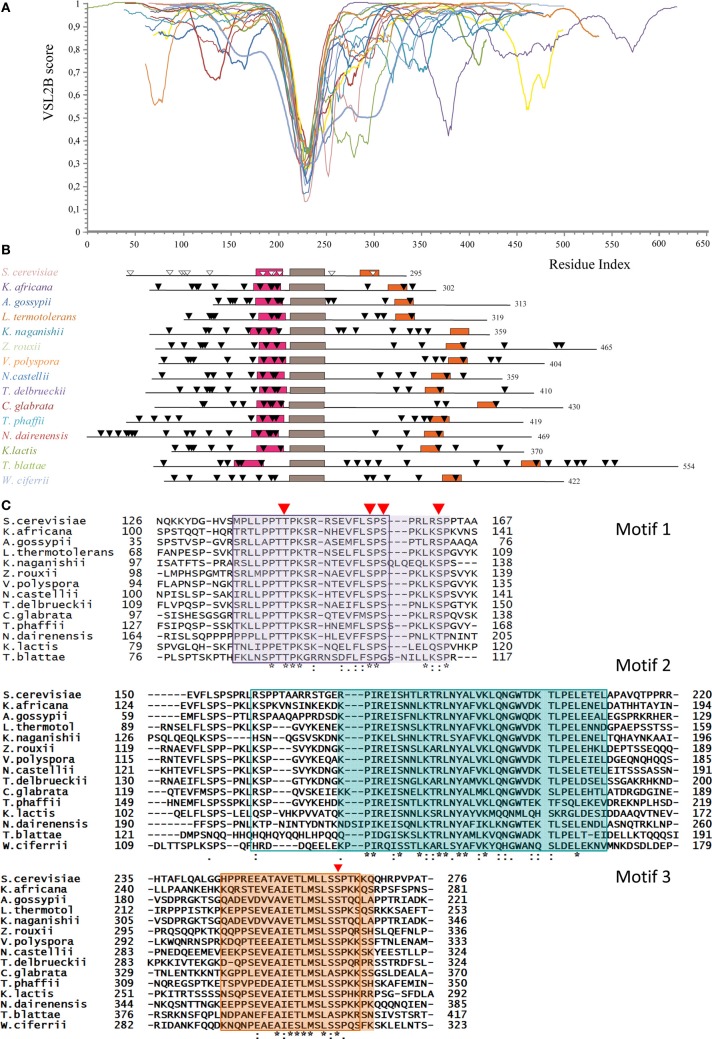
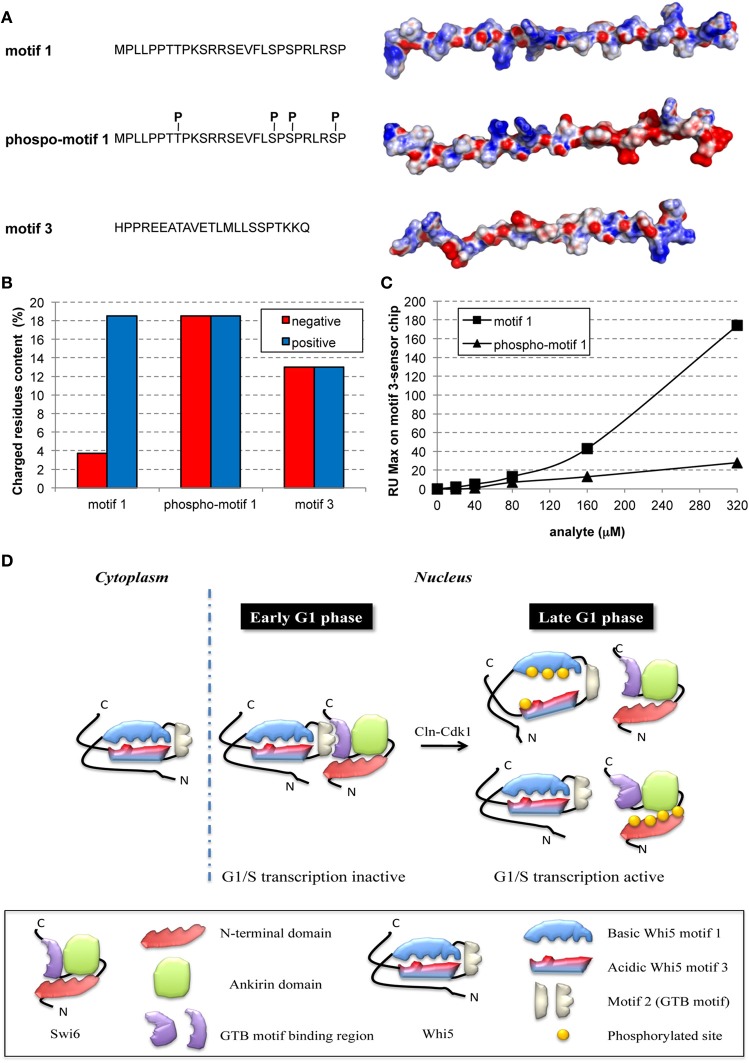
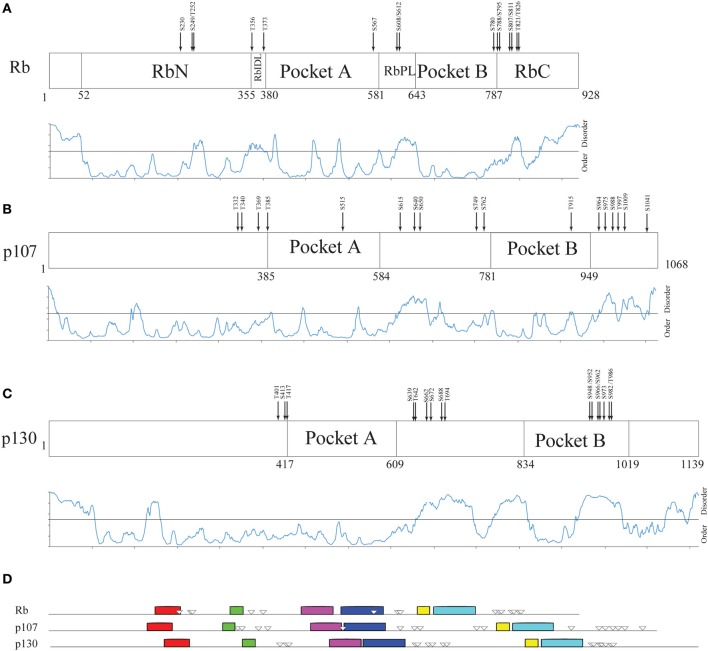
Cell growth and proliferation require a complex series of tight-regulated and well-orchestrated events. Accordingly, proteins governing such events are evolutionary conserved, even among distant organisms. By contrast, it is more singular the case of "core functions" exerted by functional analogous proteins that are not homologous and do not share any kind of structural similarity. This is the case of proteins regulating the G1/S transition in higher eukaryotes-i.e., the retinoblastoma (Rb) tumor suppressor Rb-and budding yeast, i.e., Whi5. The interaction landscape of Rb and Whi5 is quite large, with more than one hundred proteins interacting either genetically or physically with each protein. The Whi5 interactome has been used to construct a concept map of Whi5 function and regulation. Comparison of physical and genetic interactors of Rb and Whi5 allows highlighting a significant core of conserved, common functionalities associated with the interactors indicating that structure and function of the network-rather than individual proteins-are conserved during evolution. A combined bioinformatics and biochemical approach has shown that the whole Whi5 protein is highly disordered, except for a small region containing the protein family signature. The comparison with Whi5 homologs from Saccharomycetales has prompted the hypothesis of a modular organization of structural disorder, with most evolutionary conserved regions alternating with highly variable ones. The finding of a consensus sequence points to the conservation of a specific phosphorylation rhythm along with two disordered sequence motifs, probably acting as phosphorylation-dependent seeds in Whi5 folding/unfolding. Thus, the widely disordered Whi5 appears to act as a hierarchical, "date hub" that has evolutionary assayed an original way of modular organization before being supplanted by the globular, multi-domain structured Rb, more suitable to cover the role of a "party hub".
Keywords: cell cycle; date hub; multisite phosphorylation; party hub; protein evolution; protein hub; structural disorder; systems biology.
Figures






References
-
- Bailey T. L., Elkan C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36 - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases