

PI3K and cancer: lessons, challenges and opportunities
- PMID: 24481312
- PMCID: PMC3994981
- DOI: 10.1038/nrd4204
PI3K and cancer: lessons, challenges and opportunities
Abstract
The central role of phosphoinositide 3-kinase (PI3K) activation in tumour cell biology has prompted a sizeable effort to target PI3K and/or downstream kinases such as AKT and mammalian target of rapamycin (mTOR) in cancer. However, emerging clinical data show limited single-agent activity of inhibitors targeting PI3K, AKT or mTOR at tolerated doses. One exception is the response to PI3Kδ inhibitors in chronic lymphocytic leukaemia, where a combination of cell-intrinsic and -extrinsic activities drive efficacy. Here, we review key challenges and opportunities for the clinical development of inhibitors targeting the PI3K-AKT-mTOR pathway. Through a greater focus on patient selection, increased understanding of immune modulation and strategic application of rational combinations, it should be possible to realize the potential of this promising class of targeted anticancer agents.
Figures
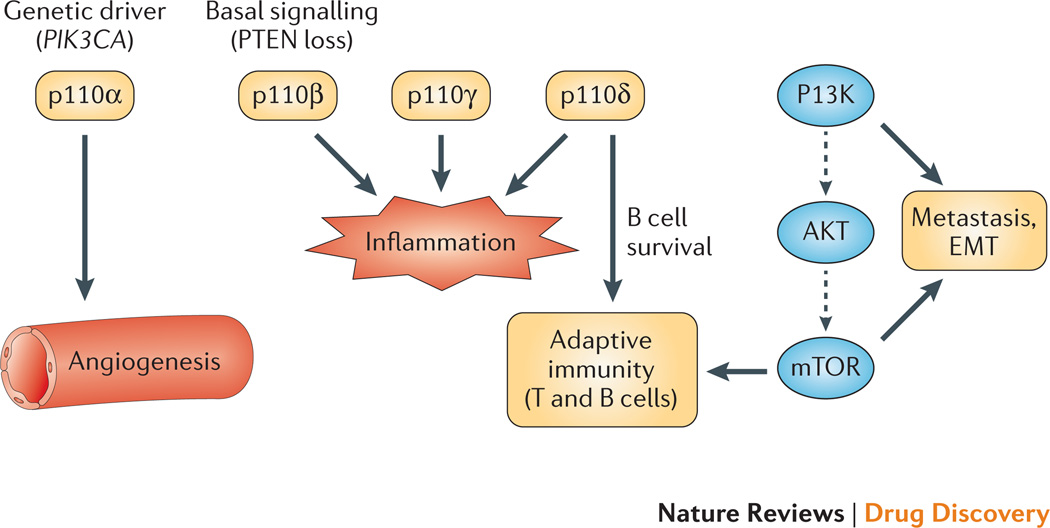
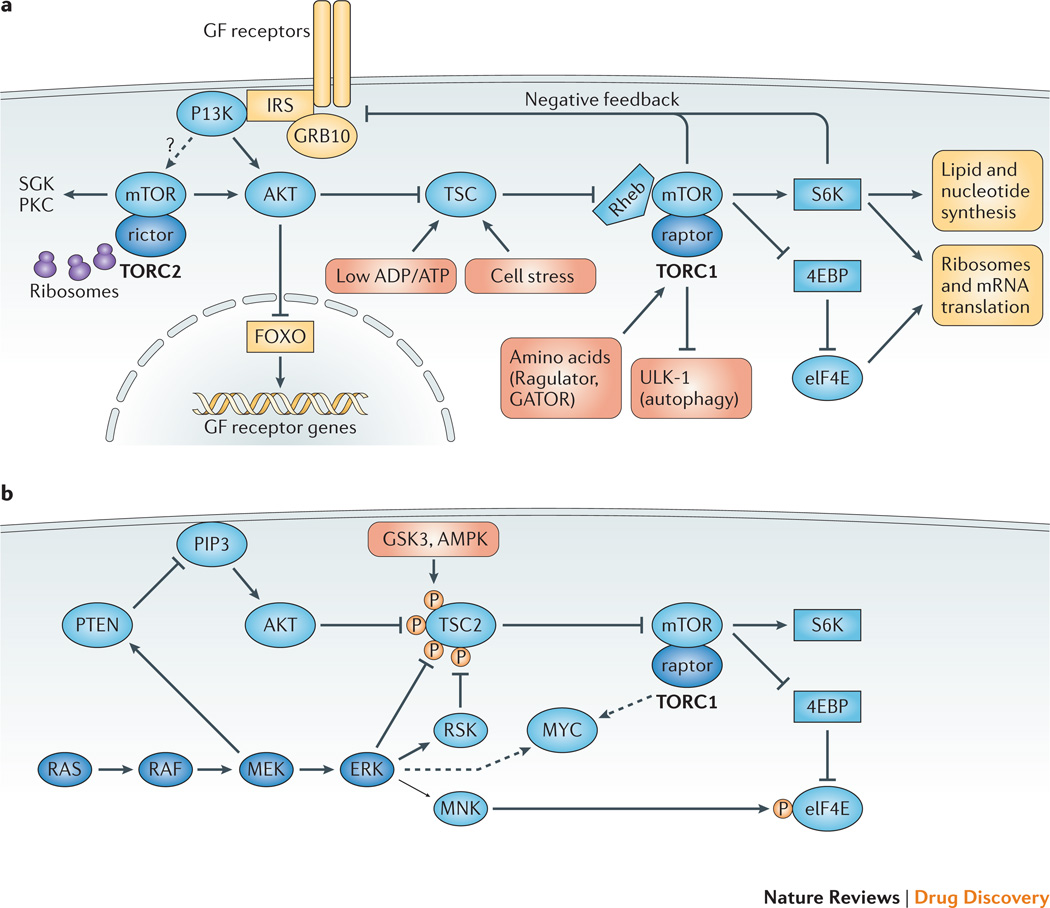
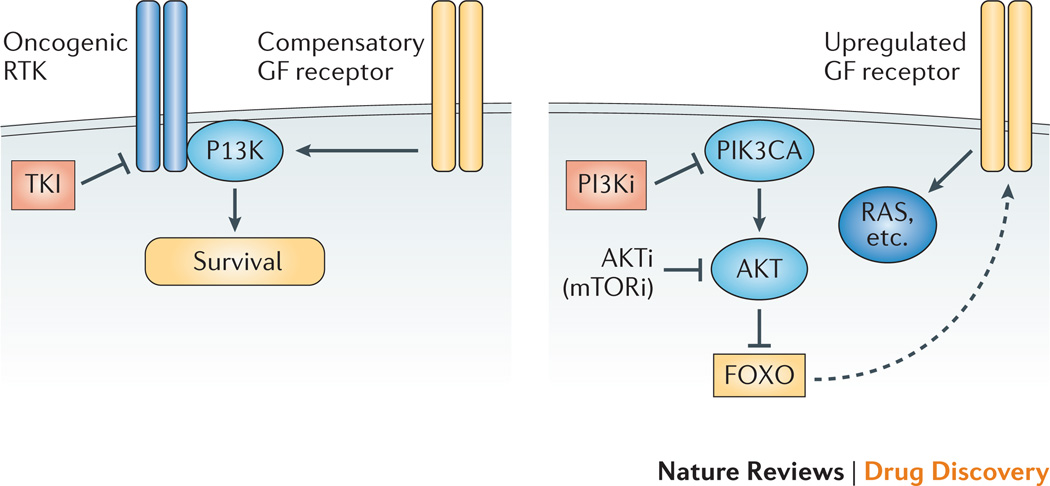
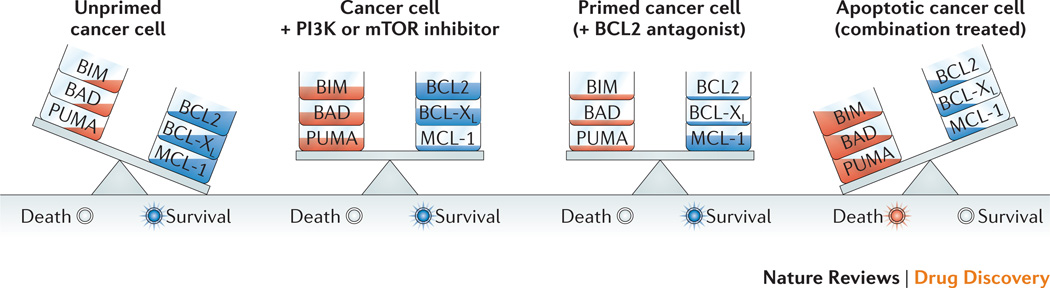
References
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. - PubMed
-
- Graupera M, Potente M. Regulation of angiogenesis by PI3K signaling networks. Exp Cell Res. 2013;319:1348–1355. - PubMed
-
- Hirsch E, Ciraolo E, Franco I, Ghigo A, Martini M. PI3K in cancer-stroma interactions: bad in seed and ugly in soil. Oncogene. 2013 Epub July 29. - PubMed
-
- Vanhaesebroeck B, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
