

Targeted metabolomics connects thioredoxin-interacting protein (TXNIP) to mitochondrial fuel selection and regulation of specific oxidoreductase enzymes in skeletal muscle
- PMID: 24482226
- PMCID: PMC3961642
- DOI: 10.1074/jbc.M113.511535
Targeted metabolomics connects thioredoxin-interacting protein (TXNIP) to mitochondrial fuel selection and regulation of specific oxidoreductase enzymes in skeletal muscle
Abstract
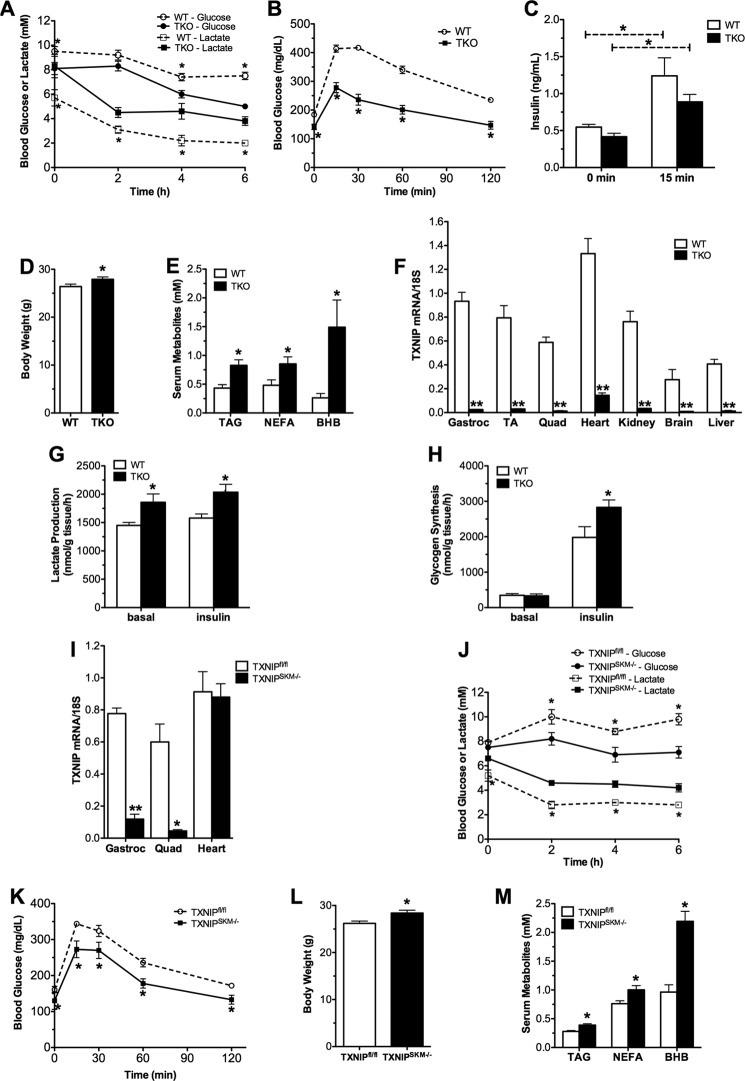
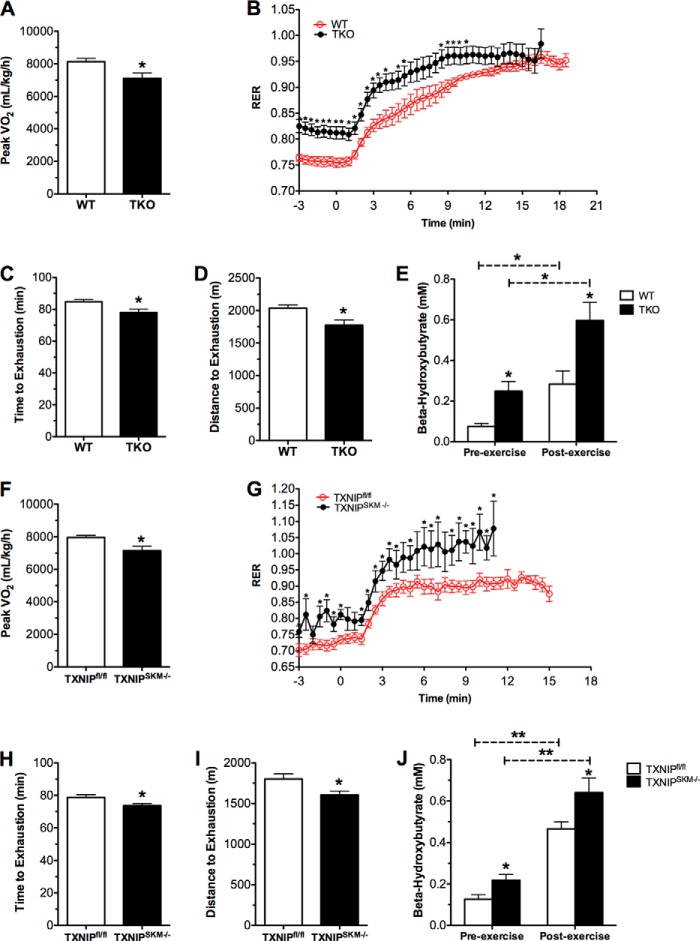
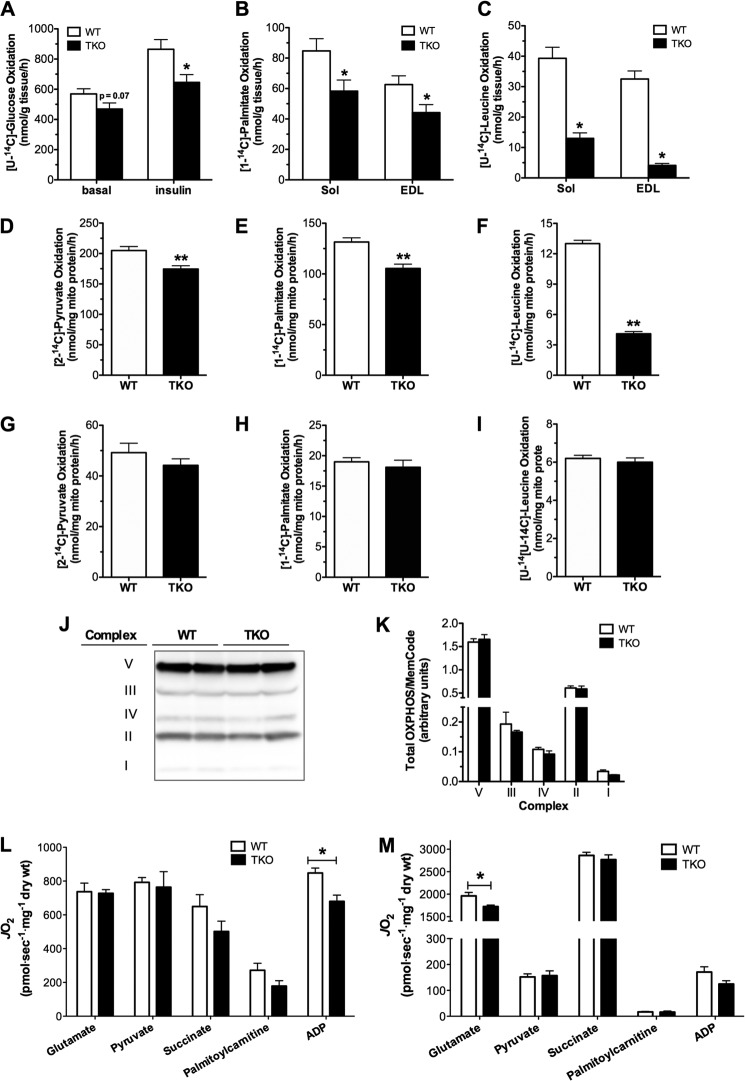
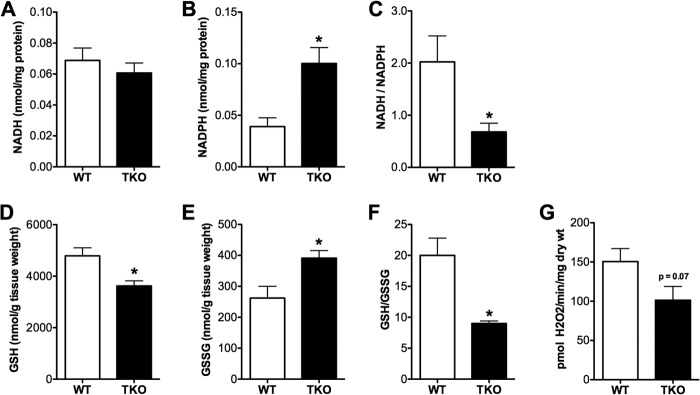
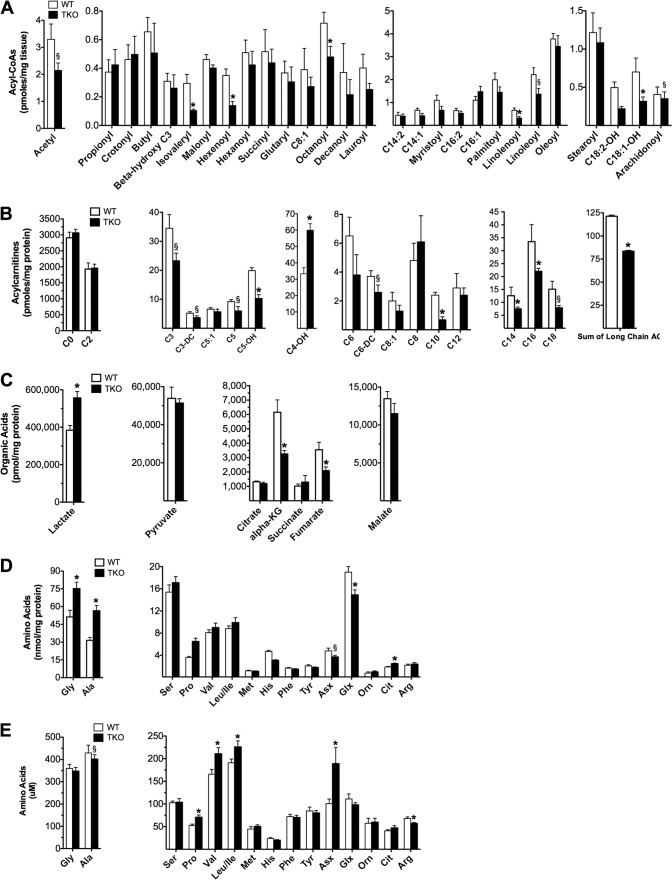
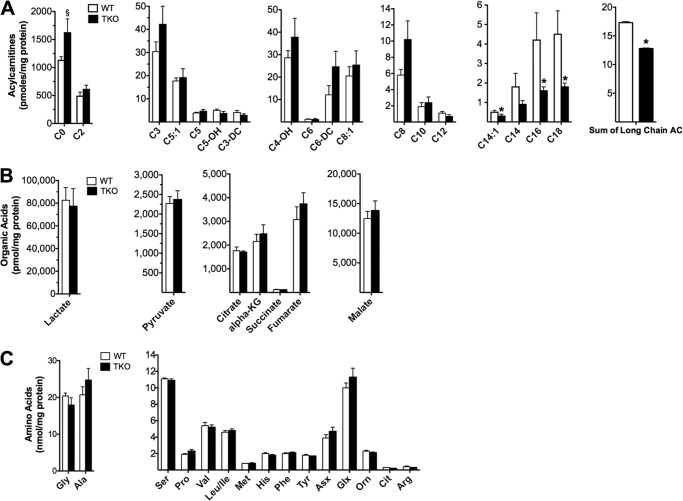
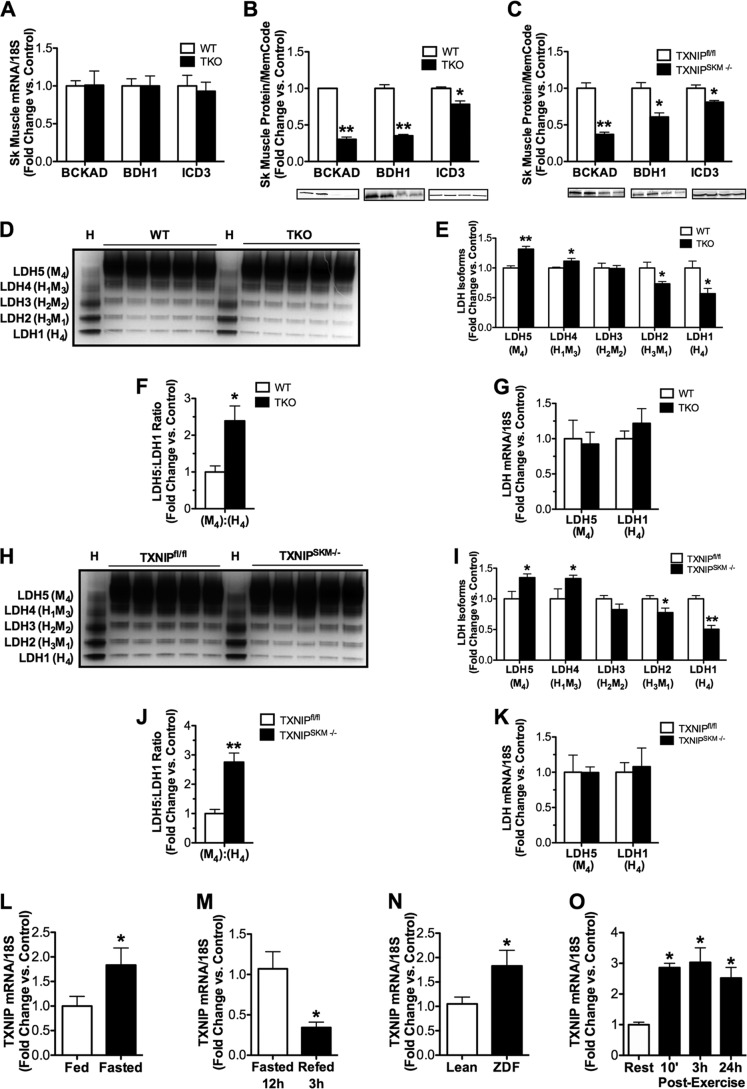
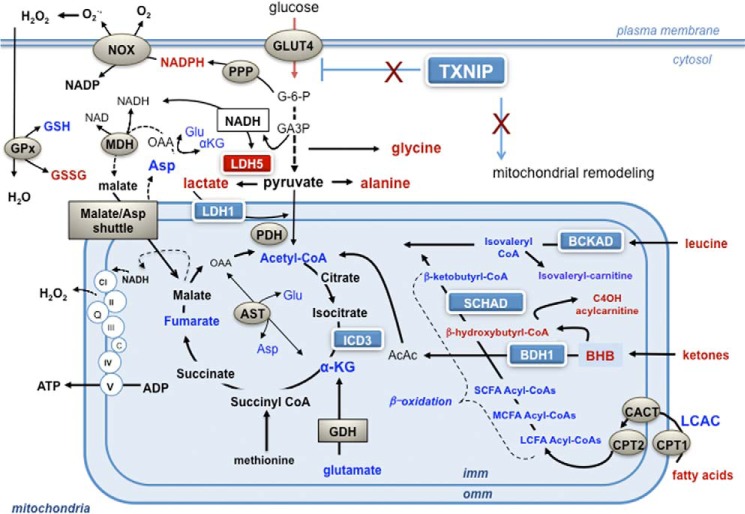
Thioredoxin-interacting protein (TXNIP) is an α-arrestin family member involved in redox sensing and metabolic control. Growing evidence links TXNIP to mitochondrial function, but the molecular nature of this relationship has remained poorly defined. Herein, we employed targeted metabolomics and comprehensive bioenergetic analyses to evaluate oxidative metabolism and respiratory kinetics in mouse models of total body (TKO) and skeletal muscle-specific (TXNIP(SKM-/-)) Txnip deficiency. Compared with littermate controls, both TKO and TXNIP(SKM-/-) mice had reduced exercise tolerance in association with muscle-specific impairments in substrate oxidation. Oxidative insufficiencies in TXNIP null muscles were not due to perturbations in mitochondrial mass, the electron transport chain, or emission of reactive oxygen species. Instead, metabolic profiling analyses led to the discovery that TXNIP deficiency causes marked deficits in enzymes required for catabolism of branched chain amino acids, ketones, and lactate, along with more modest reductions in enzymes of β-oxidation and the tricarboxylic acid cycle. The decrements in enzyme activity were accompanied by comparable deficits in protein abundance without changes in mRNA expression, implying dysregulation of protein synthesis or stability. Considering that TXNIP expression increases in response to starvation, diabetes, and exercise, these findings point to a novel role for TXNIP in coordinating mitochondrial fuel switching in response to nutrient availability.
Keywords: Branched Chain Amino Acids; Diabetes; Energy Metabolism; Ketone Body Metabolism; Mitochondrial Metabolism; Redox; Skeletal Muscle Metabolism; Thioredoxin Interacting Protein.
Figures
References
-
- Chen K. S., DeLuca H. F. (1994) Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D3. Biochim. Biophys. Acta 1219, 26–32 - PubMed
-
- Nishiyama A., Matsui M., Iwata S., Hirota K., Masutani H., Nakamura H., Takagi Y., Sono H., Gon Y., Yodoi J. (1999) Identification of thioredoxin-binding protein-2/vitamin D3 up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 274, 21645–21650 - PubMed
-
- Bodnar J. S., Chatterjee A., Castellani L. W., Ross D. A., Ohmen J., Cavalcoli J., Wu C., Dains K. M., Catanese J., Chu M., Sheth S. S., Charugundla K., Demant P., West D. B., de Jong P., Lusis A. J. (2002) Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat. Genet. 30, 110–116 - PMC - PubMed
-
- Nordberg J., Arnér E. S. (2001) Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 31, 1287–1312 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
