

Amino acid signature enables proteins to recognize modified tRNA
- PMID: 24483944
- PMCID: PMC3985708
- DOI: 10.1021/bi401174h
Amino acid signature enables proteins to recognize modified tRNA
Abstract
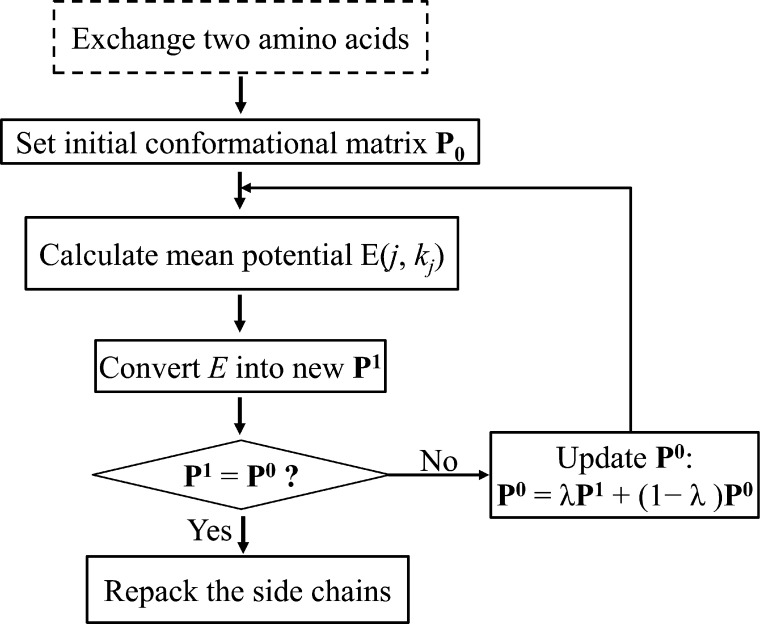
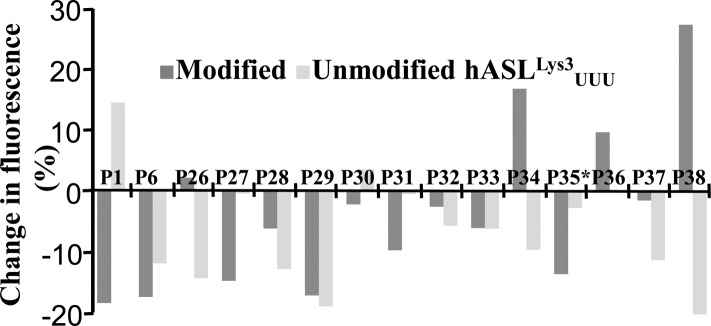
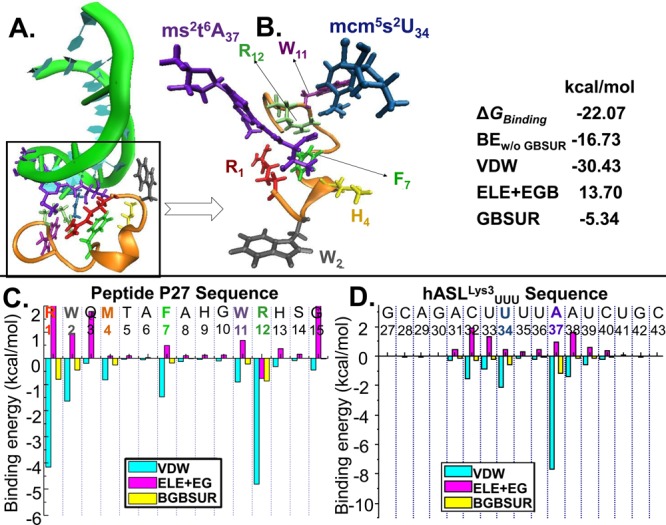
Human tRNA(Lys3)UUU is the primer for HIV replication. The HIV-1 nucleocapsid protein, NCp7, facilitates htRNA(Lys3)UUU recruitment from the host cell by binding to and remodeling the tRNA structure. Human tRNA(Lys3)UUU is post-transcriptionally modified, but until recently, the importance of those modifications in tRNA recognition by NCp7 was unknown. Modifications such as the 5-methoxycarbonylmethyl-2-thiouridine at anticodon wobble position-34 and 2-methylthio-N(6)-threonylcarbamoyladenosine, adjacent to the anticodon at position-37, are important to the recognition of htRNA(Lys3)UUU by NCp7. Several short peptides selected from phage display libraries were found to also preferentially recognize these modifications. Evolutionary algorithms (Monte Carlo and self-consistent mean field) and assisted model building with energy refinement were used to optimize the peptide sequence in silico, while fluorescence assays were developed and conducted to verify the in silico results and elucidate a 15-amino acid signature sequence (R-W-Q/N-H-X2-F-Pho-X-G/A-W-R-X2-G, where X can be most amino acids, and Pho is hydrophobic) that recognized the tRNA's fully modified anticodon stem and loop domain, hASL(Lys3)UUU. Peptides of this sequence specifically recognized and bound modified htRNA(Lys3)UUU with an affinity 10-fold higher than that of the starting sequence. Thus, this approach provides an effective means of predicting sequences of RNA binding peptides that have better binding properties. Such peptides can be used in cell and molecular biology as well as biochemistry to explore RNA binding proteins and to inhibit those protein functions.
Figures


Similar articles
-
Functional recognition of the modified human tRNALys3(UUU) anticodon domain by HIV's nucleocapsid protein and a peptide mimic.J Mol Biol. 2011 Jul 22;410(4):698-715. doi: 10.1016/j.jmb.2011.04.025. J Mol Biol. 2011. PMID: 21762809 Free PMC article.
-
Human tRNA(Lys3)(UUU) is pre-structured by natural modifications for cognate and wobble codon binding through keto-enol tautomerism.J Mol Biol. 2012 Mar 2;416(4):467-85. doi: 10.1016/j.jmb.2011.12.048. Epub 2011 Dec 29. J Mol Biol. 2012. PMID: 22227389 Free PMC article.
-
Functional anticodon architecture of human tRNALys3 includes disruption of intraloop hydrogen bonding by the naturally occurring amino acid modification, t6A.Biochemistry. 2000 Nov 7;39(44):13396-404. doi: 10.1021/bi0013039. Biochemistry. 2000. PMID: 11063577
-
Aspects of HIV-1 assembly that promote primer tRNA(Lys3) annealing to viral RNA.Virus Res. 2012 Nov;169(2):340-8. doi: 10.1016/j.virusres.2012.06.001. Epub 2012 Jun 12. Virus Res. 2012. PMID: 22698876 Review.
-
tRNA(Lys3): the primer tRNA for reverse transcription in HIV-1.IUBMB Life. 2002 Feb;53(2):107-14. doi: 10.1080/15216540211469. IUBMB Life. 2002. PMID: 12049192 Review.
Cited by
-
Computational evolution of an RNA-binding protein towards enhanced oxidized-RNA binding.Comput Struct Biotechnol J. 2019 Dec 27;18:137-152. doi: 10.1016/j.csbj.2019.12.003. eCollection 2020. Comput Struct Biotechnol J. 2019. PMID: 31988703 Free PMC article.
-
tRNA wobble modifications and protein homeostasis.Translation (Austin). 2016 Jan 28;4(1):e1143076. doi: 10.1080/21690731.2016.1143076. eCollection 2016 Jan-Jun. Translation (Austin). 2016. PMID: 27335723 Free PMC article. Review.
-
The Importance of Being Modified: The Role of RNA Modifications in Translational Fidelity.Enzymes. 2017;41:1-50. doi: 10.1016/bs.enz.2017.03.005. Epub 2017 Apr 22. Enzymes. 2017. PMID: 28601219 Free PMC article. Review.
-
Unreported intrinsic disorder in proteins: Building connections to the literature on IDPs.Intrinsically Disord Proteins. 2014 Dec 12;2(1):e970499. doi: 10.4161/21690693.2014.970499. eCollection 2014. Intrinsically Disord Proteins. 2014. PMID: 28232880 Free PMC article.
-
Adding energy minimization strategy to peptide-design algorithm enables better search for RNA-binding peptides: Redesigned λ N peptide binds boxB RNA.J Comput Chem. 2016 Oct 15;37(27):2423-35. doi: 10.1002/jcc.24466. Epub 2016 Aug 4. J Comput Chem. 2016. PMID: 27487990 Free PMC article.
References
-
- Ratner L.; Haseltine W.; Patarca R.; Livak K. L.; Starcich B.; Josephs S.; Doran D. R.; Rafalski J. A.; Whitehorn E. A.; Baumeister K.; Ifvanoff L.; Petteway S. R.; Pearson M. L.; Lautenberger J. A.; Papas T. S.; Ghrayeb J.; Chang N. T.; Gallo R. C.; Wong-Staal A. F. (1985) Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 313, 277–284. - PubMed
-
- Fassati A. (2012) Multiple roles of the capsid protein in the early steps of HIV-1 infection. Virus Res. 170, 15–24. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
