

Targeting DNA damage response in cancer therapy
- PMID: 24484288
- PMCID: PMC4317796
- DOI: 10.1111/cas.12366
Targeting DNA damage response in cancer therapy
Abstract
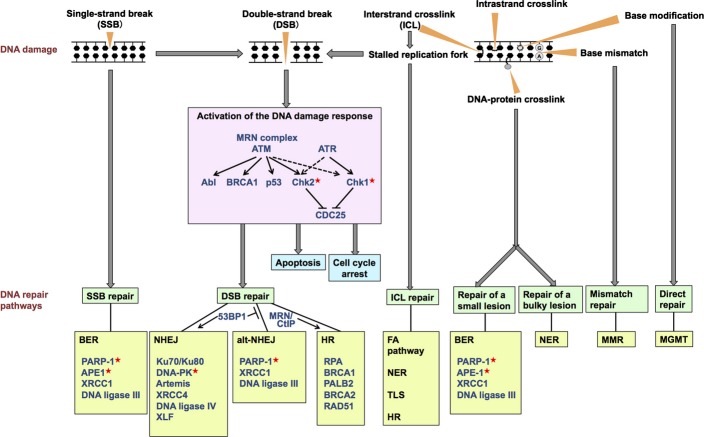
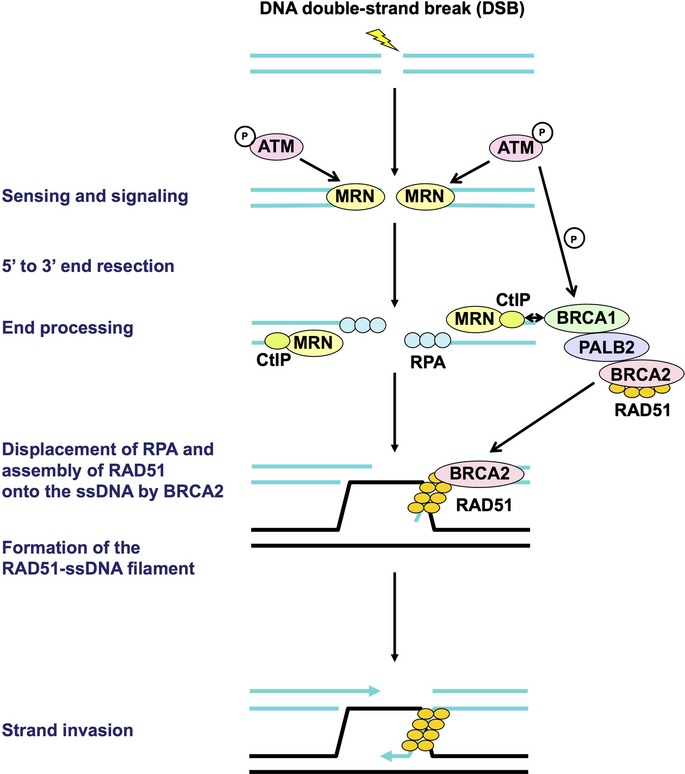
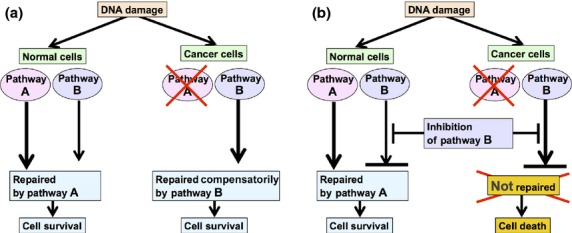
Cancer chemotherapy and radiotherapy are designed to kill cancer cells mostly by inducing DNA damage. DNA damage is normally recognized and repaired by the intrinsic DNA damage response machinery. If the damaged lesions are successfully repaired, the cells will survive. In order to specifically and effectively kill cancer cells by therapies that induce DNA damage, it is important to take advantage of specific abnormalities in the DNA damage response machinery that are present in cancer cells but not in normal cells. Such properties of cancer cells can provide biomarkers or targets for sensitization. For example, defects or upregulation of the specific pathways that recognize or repair specific types of DNA damage can serve as biomarkers of favorable or poor response to therapies that induce such types of DNA damage. Inhibition of a DNA damage response pathway may enhance the therapeutic effects in combination with the DNA-damaging agents. Moreover, it may also be useful as a monotherapy when it achieves synthetic lethality, in which inhibition of a complementary DNA damage response pathway selectively kills cancer cells that have a defect in a particular DNA repair pathway. The most striking application of this strategy is the treatment of cancers deficient in homologous recombination by poly(ADP-ribose) polymerase inhibitors. In this review, we describe the impact of targeting the cancer-specific aberrations in the DNA damage response by explaining how these treatment strategies are currently being evaluated in preclinical or clinical trials.
Keywords: Cancer therapy; DNA damage response; DNA repair; PARP inhibitors; synthetic lethality.
© 2014 The Authors. Cancer Science published by Wiley Publishing Asia Pty Ltd on behalf of Japanese Cancer Association.
Figures
References
-
- Hoeijmakers JHJ. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–85. - PubMed
-
- Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–90. - PubMed
-
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–4. - PubMed
-
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
