

RNA sequencing read depth requirement for optimal transcriptome coverage in Hevea brasiliensis
- PMID: 24484543
- PMCID: PMC3926681
- DOI: 10.1186/1756-0500-7-69
RNA sequencing read depth requirement for optimal transcriptome coverage in Hevea brasiliensis
Abstract
Background: One of the concerns of assembling de novo transcriptomes is determining the amount of read sequences required to ensure a comprehensive coverage of genes expressed in a particular sample. In this report, we describe the use of Illumina paired-end RNA-Seq (PE RNA-Seq) reads from Hevea brasiliensis (rubber tree) bark to devise a transcript mapping approach for the estimation of the read amount needed for deep transcriptome coverage.
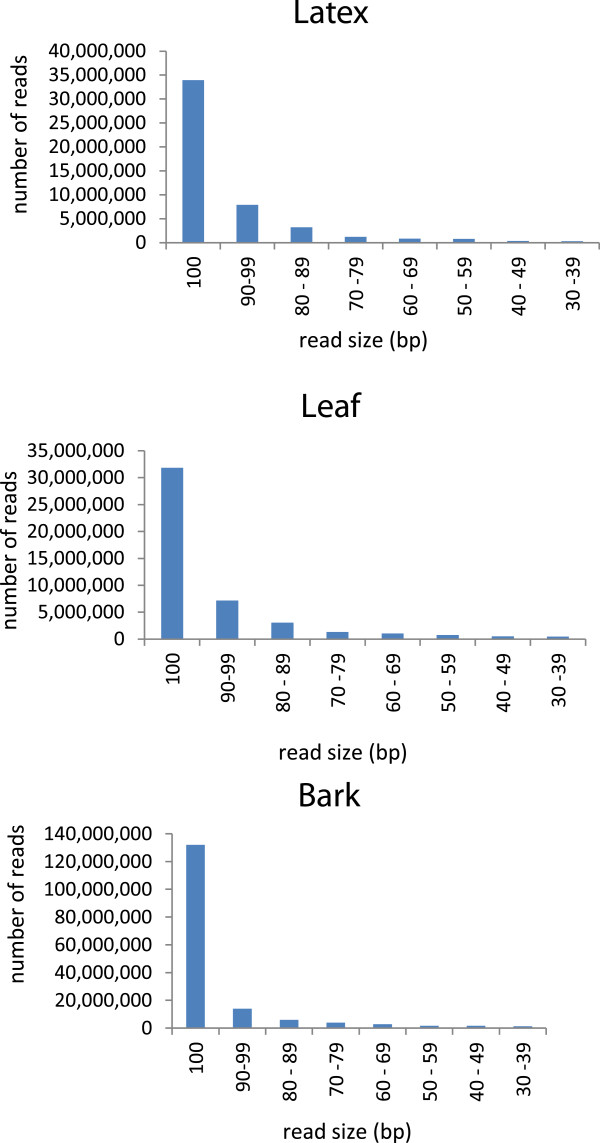
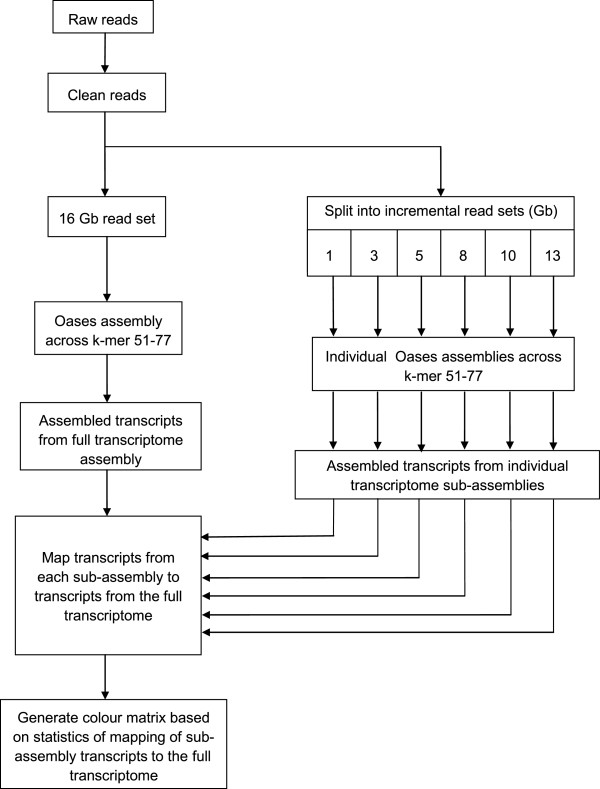
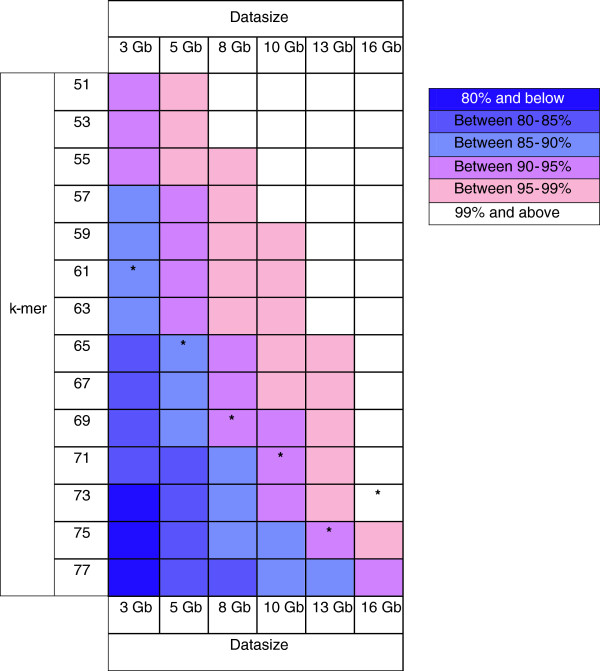
Findings: We optimized the assembly of a Hevea bark transcriptome based on 16 Gb Illumina PE RNA-Seq reads using the Oases assembler across a range of k-mer sizes. We then assessed assembly quality based on transcript N50 length and transcript mapping statistics in relation to (a) known Hevea cDNAs with complete open reading frames, (b) a set of core eukaryotic genes and (c) Hevea genome scaffolds. This was followed by a systematic transcript mapping process where sub-assemblies from a series of incremental amounts of bark transcripts were aligned to transcripts from the entire bark transcriptome assembly. The exercise served to relate read amounts to the degree of transcript mapping level, the latter being an indicator of the coverage of gene transcripts expressed in the sample. As read amounts or datasize increased toward 16 Gb, the number of transcripts mapped to the entire bark assembly approached saturation. A colour matrix was subsequently generated to illustrate sequencing depth requirement in relation to the degree of coverage of total sample transcripts.
Conclusions: We devised a procedure, the "transcript mapping saturation test", to estimate the amount of RNA-Seq reads needed for deep coverage of transcriptomes. For Hevea de novo assembly, we propose generating between 5-8 Gb reads, whereby around 90% transcript coverage could be achieved with optimized k-mers and transcript N50 length. The principle behind this methodology may also be applied to other non-model plants, or with reads from other second generation sequencing platforms.
Figures

Similar articles
-
De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.).BMC Genomics. 2012 May 18;13:192. doi: 10.1186/1471-2164-13-192. BMC Genomics. 2012. PMID: 22607098 Free PMC article.
-
Molecular mechanism of ethylene stimulation of latex yield in rubber tree (Hevea brasiliensis) revealed by de novo sequencing and transcriptome analysis.BMC Genomics. 2016 Mar 24;17:257. doi: 10.1186/s12864-016-2587-4. BMC Genomics. 2016. PMID: 27008913 Free PMC article.
-
RNA-Seq analysis and de novo transcriptome assembly of Hevea brasiliensis.Plant Mol Biol. 2011 Oct;77(3):299-308. doi: 10.1007/s11103-011-9811-z. Epub 2011 Aug 3. Plant Mol Biol. 2011. PMID: 21811850
-
Strategies for transcriptome analysis in nonmodel plants.Am J Bot. 2012 Feb;99(2):267-76. doi: 10.3732/ajb.1100334. Epub 2012 Feb 1. Am J Bot. 2012. PMID: 22301897 Review.
-
Genomic technologies for Hevea breeding.Adv Genet. 2019;104:1-73. doi: 10.1016/bs.adgen.2019.04.001. Epub 2019 Jun 3. Adv Genet. 2019. PMID: 31200808 Review.
Cited by
-
Transcriptome Analysis of the Signalling Networks in Coronatine-Induced Secondary Laticifer Differentiation from Vascular Cambia in Rubber Trees.Sci Rep. 2016 Nov 3;6:36384. doi: 10.1038/srep36384. Sci Rep. 2016. PMID: 27808245 Free PMC article.
-
RNA-seq analysis and de novo transcriptome assembly of Jerusalem artichoke (Helianthus tuberosus Linne).PLoS One. 2014 Nov 6;9(11):e111982. doi: 10.1371/journal.pone.0111982. eCollection 2014. PLoS One. 2014. PMID: 25375764 Free PMC article.
-
Regulation of HbPIP2;3, a Latex-Abundant Water Transporter, Is Associated with Latex Dilution and Yield in the Rubber Tree (Hevea brasiliensis Muell. Arg.).PLoS One. 2015 Apr 30;10(4):e0125595. doi: 10.1371/journal.pone.0125595. eCollection 2015. PLoS One. 2015. PMID: 25927524 Free PMC article.
-
Comparative transcriptome analysis of latex from rubber tree clone CATAS8-79 and PR107 reveals new cues for the regulation of latex regeneration and duration of latex flow.BMC Plant Biol. 2015 Apr 18;15:104. doi: 10.1186/s12870-015-0488-3. BMC Plant Biol. 2015. PMID: 25928745 Free PMC article.
-
Optimal sequencing depth design for whole genome re-sequencing in pigs.BMC Bioinformatics. 2019 Nov 8;20(1):556. doi: 10.1186/s12859-019-3164-z. BMC Bioinformatics. 2019. PMID: 31703550 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
