

A general framework for estimating the relative pathogenicity of human genetic variants
- PMID: 24487276
- PMCID: PMC3992975
- DOI: 10.1038/ng.2892
A general framework for estimating the relative pathogenicity of human genetic variants
Abstract
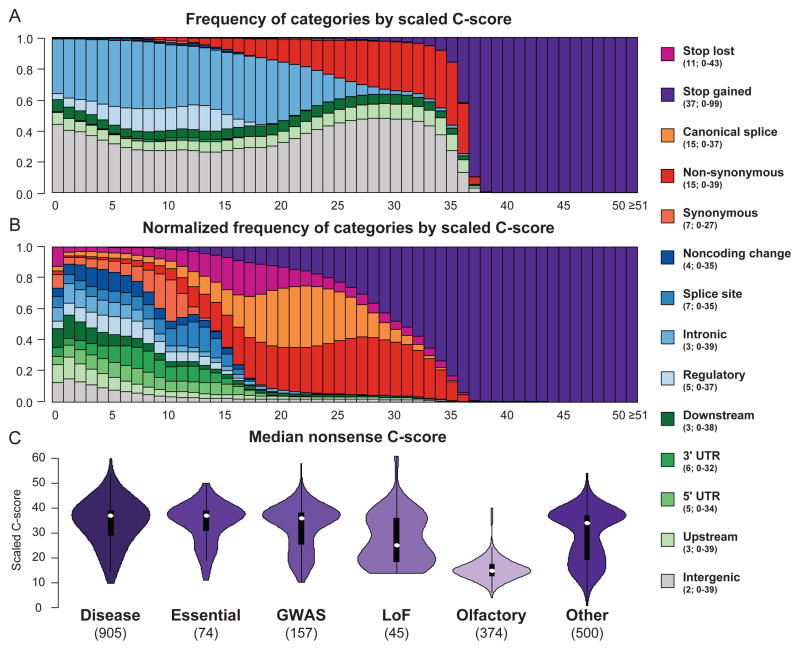
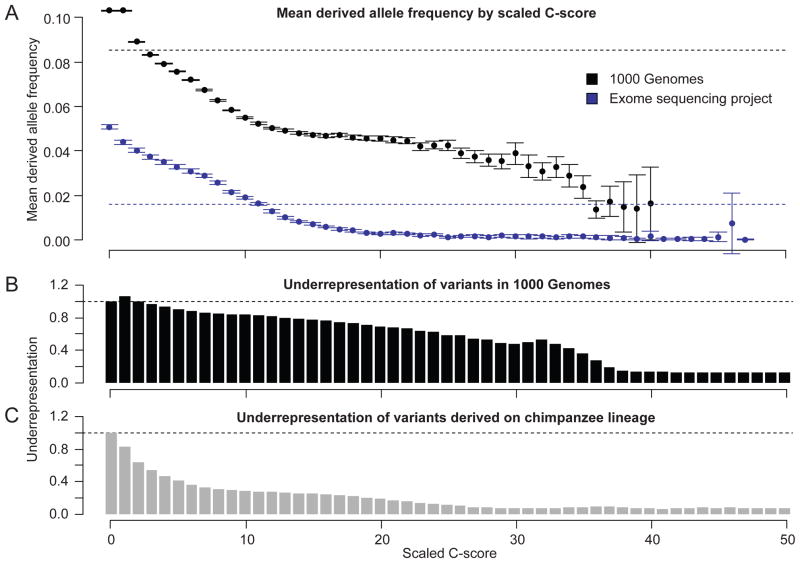
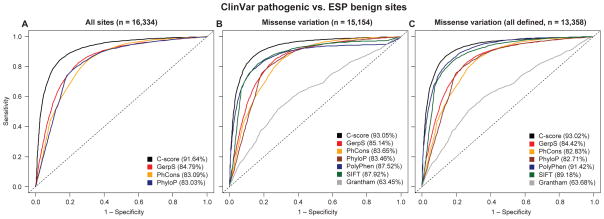
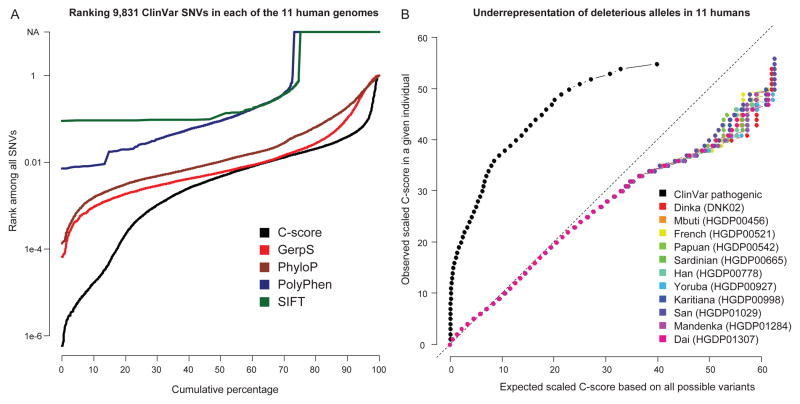
Current methods for annotating and interpreting human genetic variation tend to exploit a single information type (for example, conservation) and/or are restricted in scope (for example, to missense changes). Here we describe Combined Annotation-Dependent Depletion (CADD), a method for objectively integrating many diverse annotations into a single measure (C score) for each variant. We implement CADD as a support vector machine trained to differentiate 14.7 million high-frequency human-derived alleles from 14.7 million simulated variants. We precompute C scores for all 8.6 billion possible human single-nucleotide variants and enable scoring of short insertions-deletions. C scores correlate with allelic diversity, annotations of functionality, pathogenicity, disease severity, experimentally measured regulatory effects and complex trait associations, and they highly rank known pathogenic variants within individual genomes. The ability of CADD to prioritize functional, deleterious and pathogenic variants across many functional categories, effect sizes and genetic architectures is unmatched by any current single-annotation method.
Figures
Comment in
-
Disease genetics: all together now for variant interpretation.Nat Rev Genet. 2014 Apr;15(4):216. doi: 10.1038/nrg3702. Epub 2014 Feb 18. Nat Rev Genet. 2014. PMID: 24535247 No abstract available.
References
-
- Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet. 2011;12:628–40. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
