

In vivo mapping of arabidopsis scaffold/matrix attachment regions reveals link to nucleosome-disfavoring poly(dA:dT) tracts
- PMID: 24488963
- PMCID: PMC3963562
- DOI: 10.1105/tpc.113.121194
In vivo mapping of arabidopsis scaffold/matrix attachment regions reveals link to nucleosome-disfavoring poly(dA:dT) tracts
Abstract
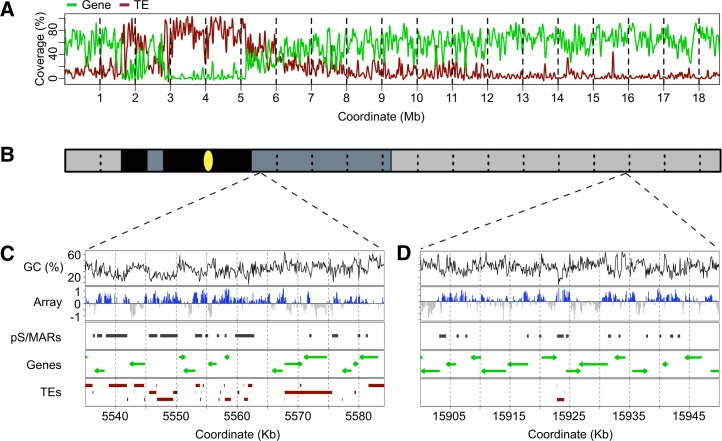
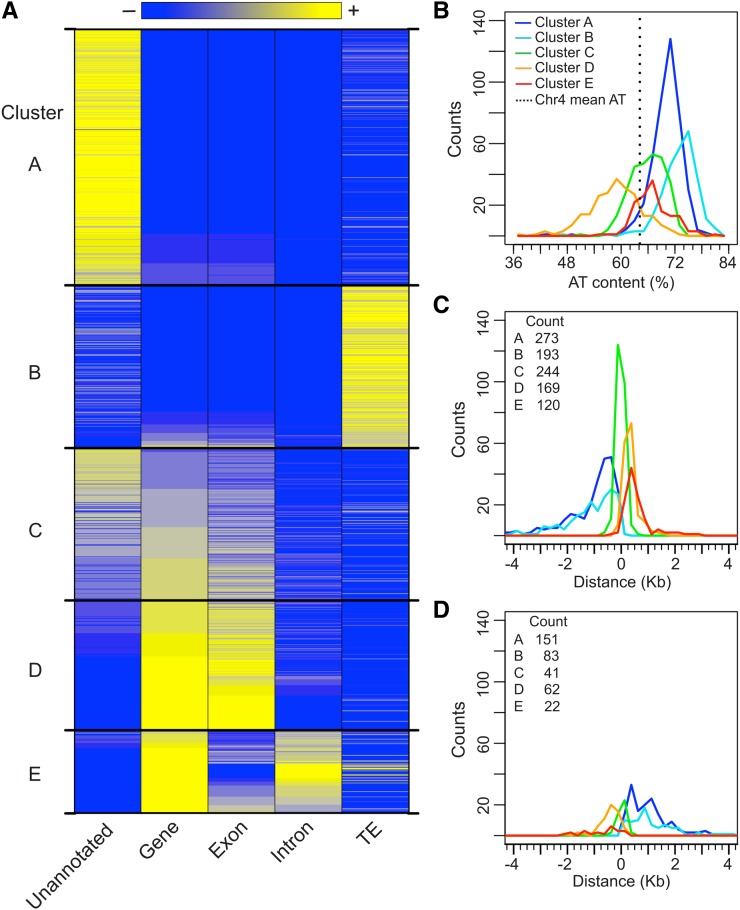
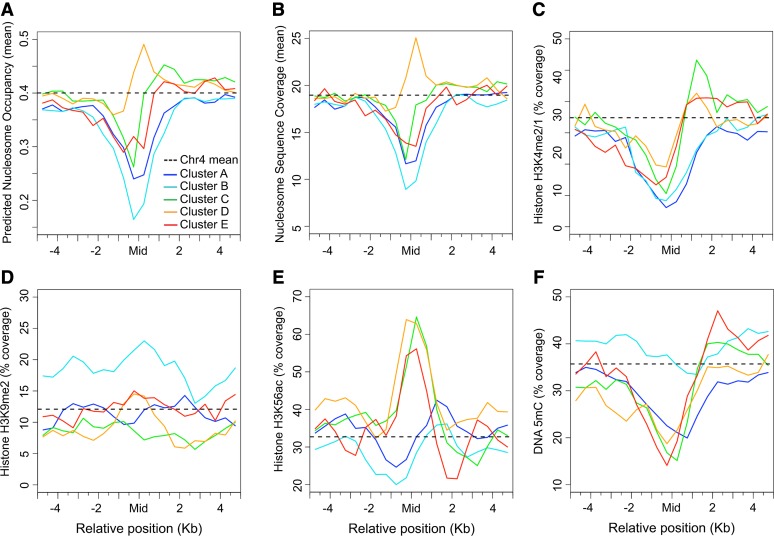
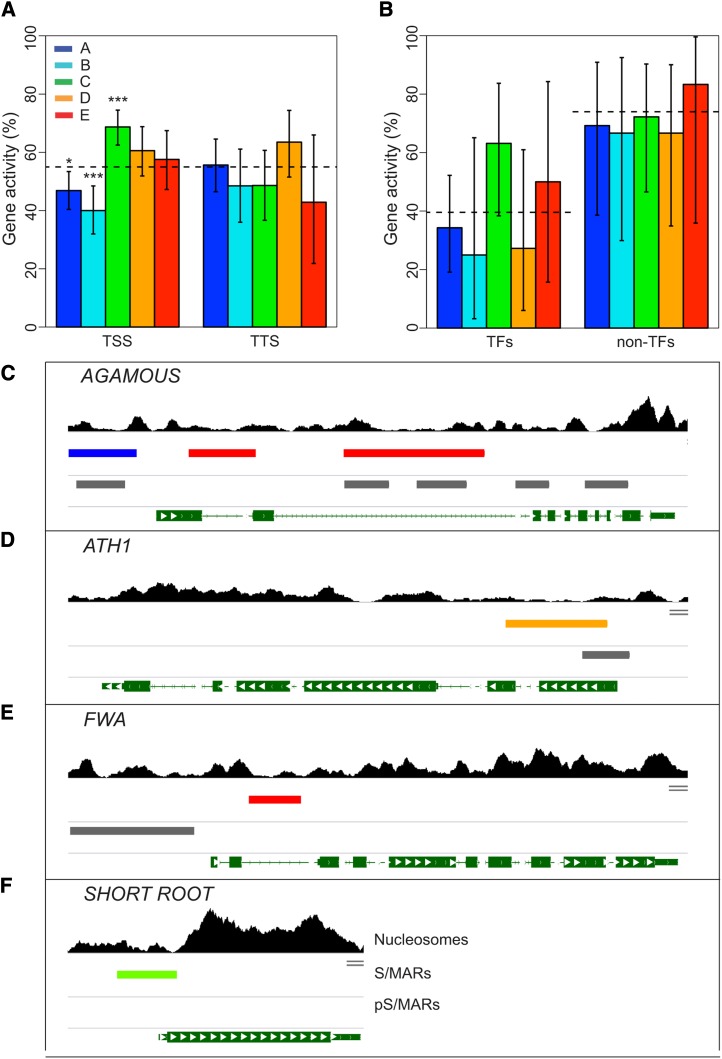
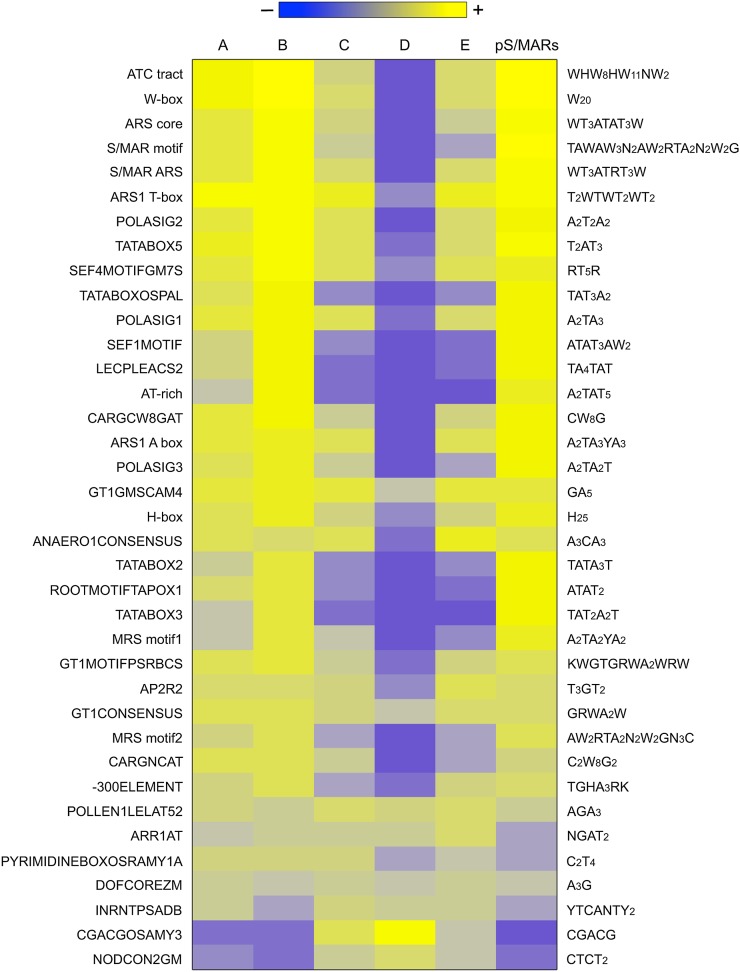
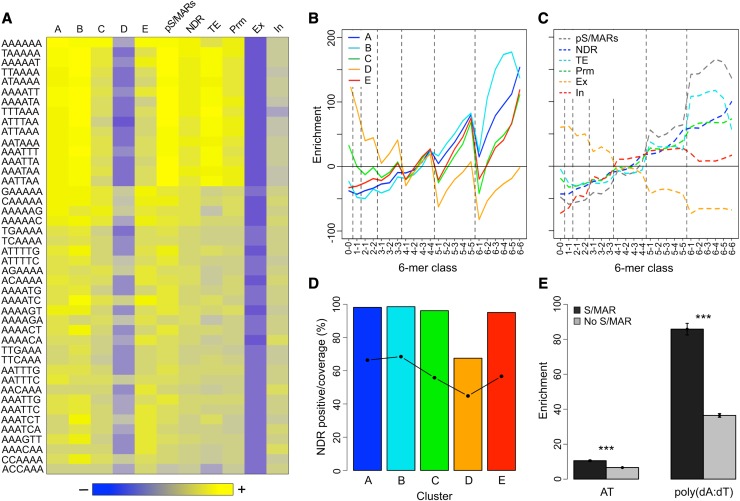
Scaffold or matrix attachment regions (S/MARs) are found in all eukaryotes. The pattern of distribution and genomic context of S/MARs is thought to be important for processes such as chromatin organization and modulation of gene expression. Despite the importance of such processes, much is unknown about the large-scale distribution and sequence content of S/MARs in vivo. Here, we report the use of tiling microarrays to map 1358 S/MARs on Arabidopsis thaliana chromosome 4 (chr4). S/MARs occur throughout chr4, spaced much more closely than in the large plant and animal genomes that have been studied to date. Arabidopsis S/MARs can be divided into five clusters based on their association with other genomic features, suggesting a diversity of functions. While some Arabidopsis S/MARs may define structural domains, most occur near the transcription start sites of genes. Genes associated with these S/MARs have an increased probability of expression, which is particularly pronounced in the case of transcription factor genes. Analysis of sequence motifs and 6-mer enrichment patterns show that S/MARs are preferentially enriched in poly(dA:dT) tracts, sequences that resist nucleosome formation, and the majority of S/MARs contain at least one nucleosome-depleted region. This global view of S/MARs provides a framework to begin evaluating genome-scale models for S/MAR function.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
