

Differential methylation during maize leaf growth targets developmentally regulated genes
- PMID: 24488968
- PMCID: PMC3938625
- DOI: 10.1104/pp.113.233312
Differential methylation during maize leaf growth targets developmentally regulated genes
Abstract
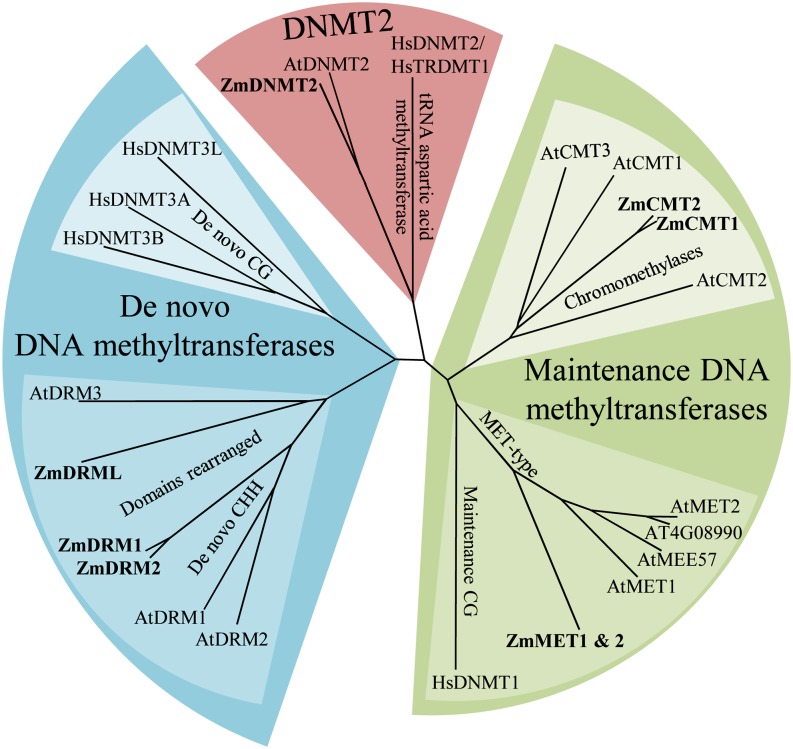
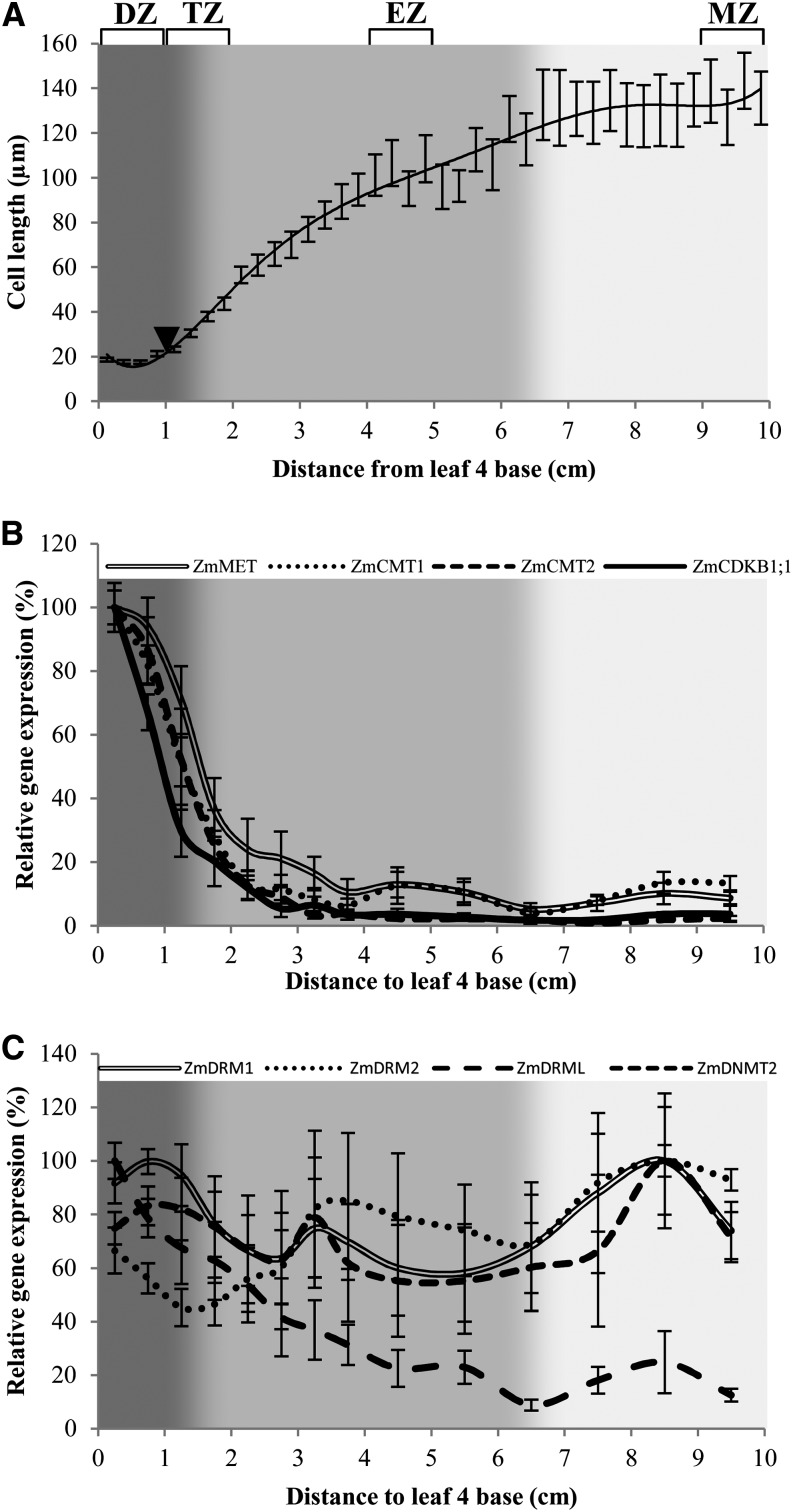
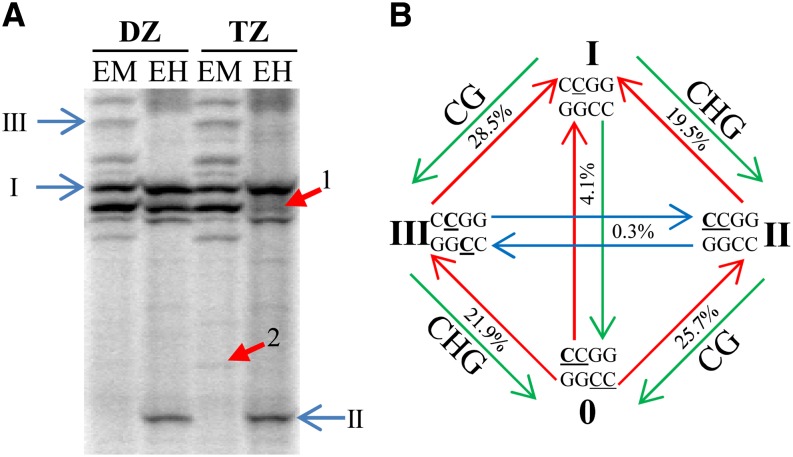
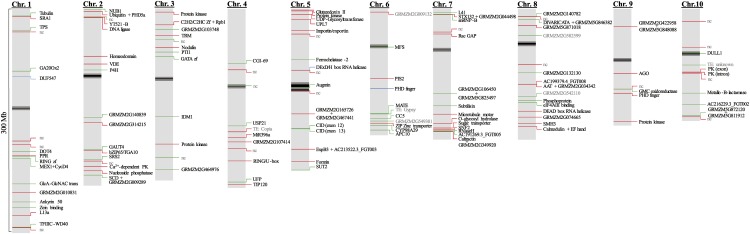
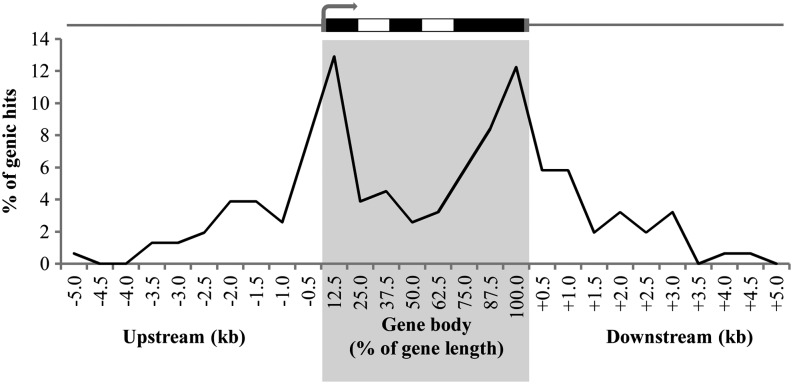
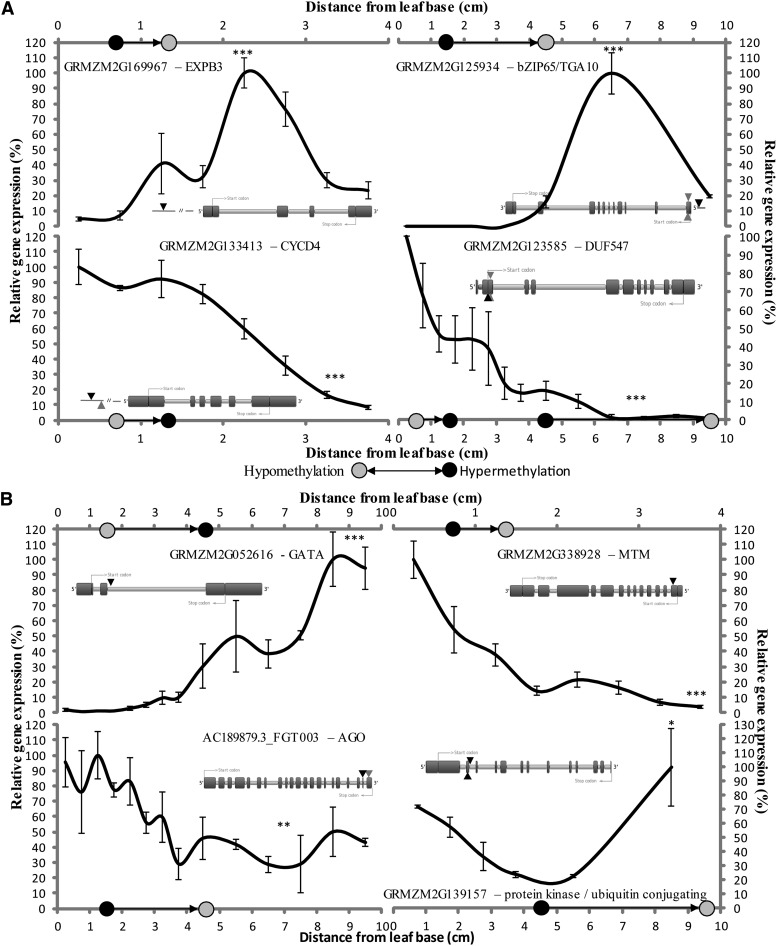
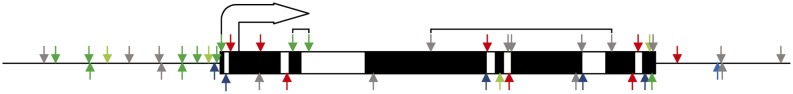
DNA methylation is an important and widespread epigenetic modification in plant genomes, mediated by DNA methyltransferases (DMTs). DNA methylation is known to play a role in genome protection, regulation of gene expression, and splicing and was previously associated with major developmental reprogramming in plants, such as vernalization and transition to flowering. Here, we show that DNA methylation also controls the growth processes of cell division and cell expansion within a growing organ. The maize (Zea mays) leaf offers a great tool to study growth processes, as the cells progressively move through the spatial gradient encompassing the division zone, transition zone, elongation zone, and mature zone. Opposite to de novo DMTs, the maintenance DMTs were transcriptionally regulated throughout the growth zone of the maize leaf, concomitant with differential CCGG methylation levels in the four zones. Surprisingly, the majority of differentially methylated sequences mapped on or close to gene bodies and not to repeat-rich loci. Moreover, especially the 5' and 3' regions of genes, which show overall low methylation levels, underwent differential methylation in a developmental context. Genes involved in processes such as chromatin remodeling, cell cycle progression, and growth regulation, were differentially methylated. The presence of differential methylation located upstream of the gene anticorrelated with transcript expression, while gene body differential methylation was unrelated to the expression level. These data indicate that DNA methylation is correlated with the decision to exit mitotic cell division and to enter cell expansion, which adds a new epigenetic level to the regulation of growth processes.
Figures

References
-
- Andriankaja M, Dhondt S, De Bodt S, Vanhaeren H, Coppens F, De Milde L, Mühlenbock P, Skirycz A, Gonzalez N, Beemster GTS, et al. (2012) Exit from proliferation during leaf development in Arabidopsis thaliana: a not-so-gradual process. Dev Cell 22: 64–78 - PubMed
-
- Ayliffe MA, Scott NS, Timmis JN. (1998) Analysis of plastid DNA-like sequences within the nuclear genomes of higher plants. Mol Biol Evol 15: 738–745 - PubMed
-
- Cervera MT, Ruiz-García L, Martínez-Zapater JM. (2002) Analysis of DNA methylation in Arabidopsis thaliana based on methylation-sensitive AFLP markers. Mol Genet Genomics 268: 543–552 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
