

A preliminary study of viral metagenomics of French bat species in contact with humans: identification of new mammalian viruses
- PMID: 24489870
- PMCID: PMC3906132
- DOI: 10.1371/journal.pone.0087194
A preliminary study of viral metagenomics of French bat species in contact with humans: identification of new mammalian viruses
Abstract
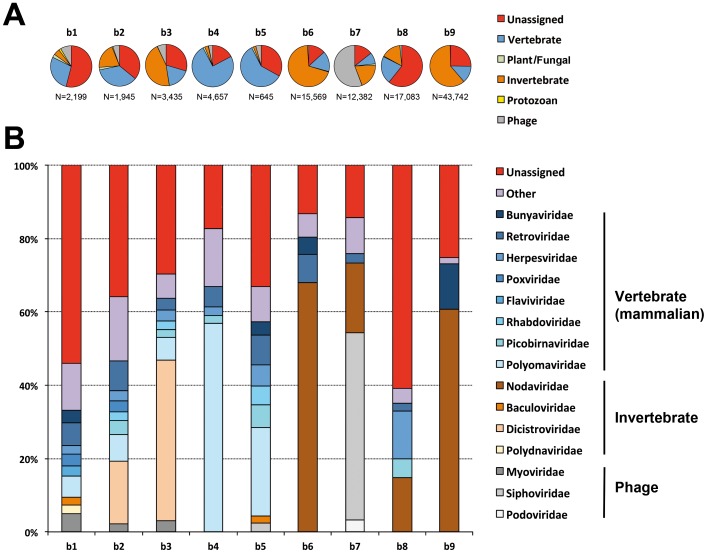
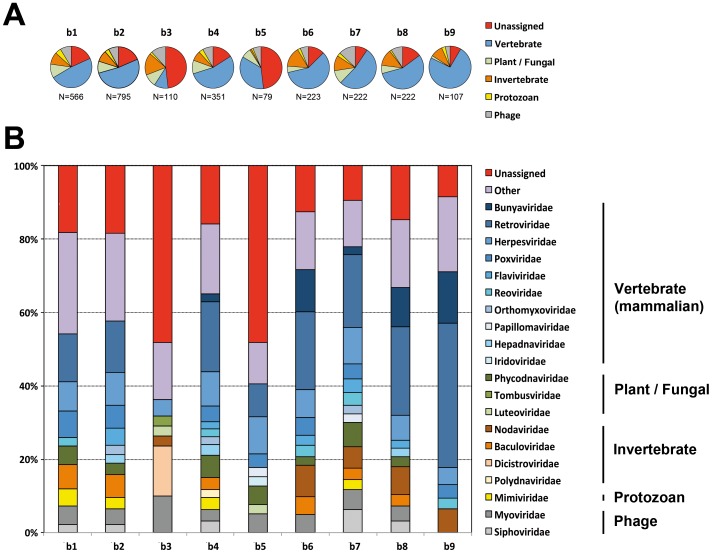
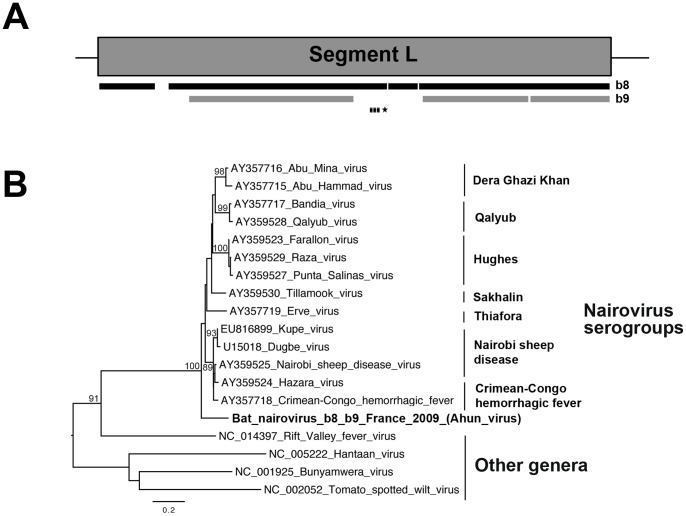
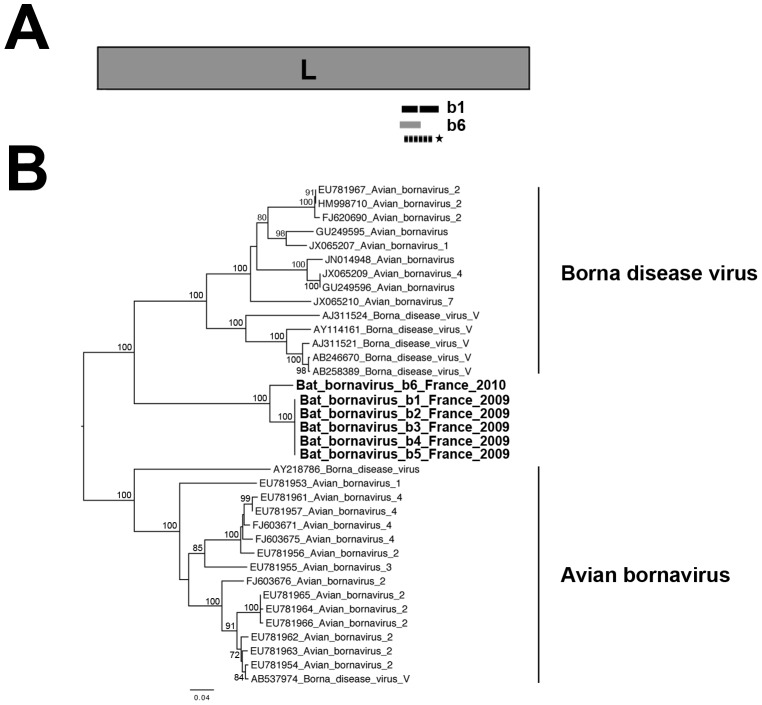
The prediction of viral zoonosis epidemics has become a major public health issue. A profound understanding of the viral population in key animal species acting as reservoirs represents an important step towards this goal. Bats harbor diverse viruses, some of which are of particular interest because they cause severe human diseases. However, little is known about the diversity of the global population of viruses found in bats (virome). We determined the viral diversity of five different French insectivorous bat species (nine specimens in total) in close contact with humans. Sequence-independent amplification, high-throughput sequencing with Illumina technology and a dedicated bioinformatics analysis pipeline were used on pooled tissues (brain, liver and lungs). Comparisons of the sequences of contigs and unassembled reads provided a global taxonomic distribution of virus-related sequences for each sample, highlighting differences both within and between bat species. Many viral families were present in these viromes, including viruses known to infect bacteria, plants/fungi, insects or vertebrates, the most relevant being those infecting mammals (Retroviridae, Herpesviridae, Bunyaviridae, Poxviridae, Flaviviridae, Reoviridae, Bornaviridae, Picobirnaviridae). In particular, we detected several new mammalian viruses, including rotaviruses, gammaretroviruses, bornaviruses and bunyaviruses with the identification of the first bat nairovirus. These observations demonstrate that bats naturally harbor viruses from many different families, most of which infect mammals. They may therefore constitute a major reservoir of viral diversity that should be analyzed carefully, to determine the role played by bats in the spread of zoonotic viral infections.
Conflict of interest statement
Figures


References
-
- van der Poel WH, Lina PH, Kramps JA (2006) Public health awareness of emerging zoonotic viruses of bats: a European perspective. Vector Borne Zoonotic Dis 6: 315–324. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
