

Macrophage migration inhibitory factor is a novel determinant of cigarette smoke-induced lung damage
- PMID: 24490973
- PMCID: PMC4091857
- DOI: 10.1165/rcmb.2013-0371OC
Macrophage migration inhibitory factor is a novel determinant of cigarette smoke-induced lung damage
Abstract
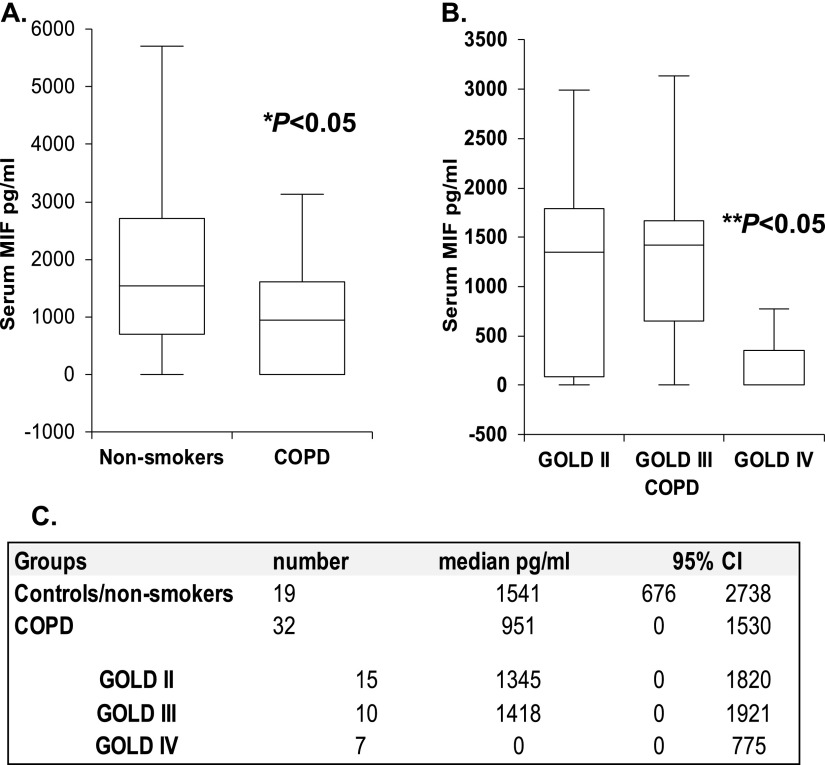
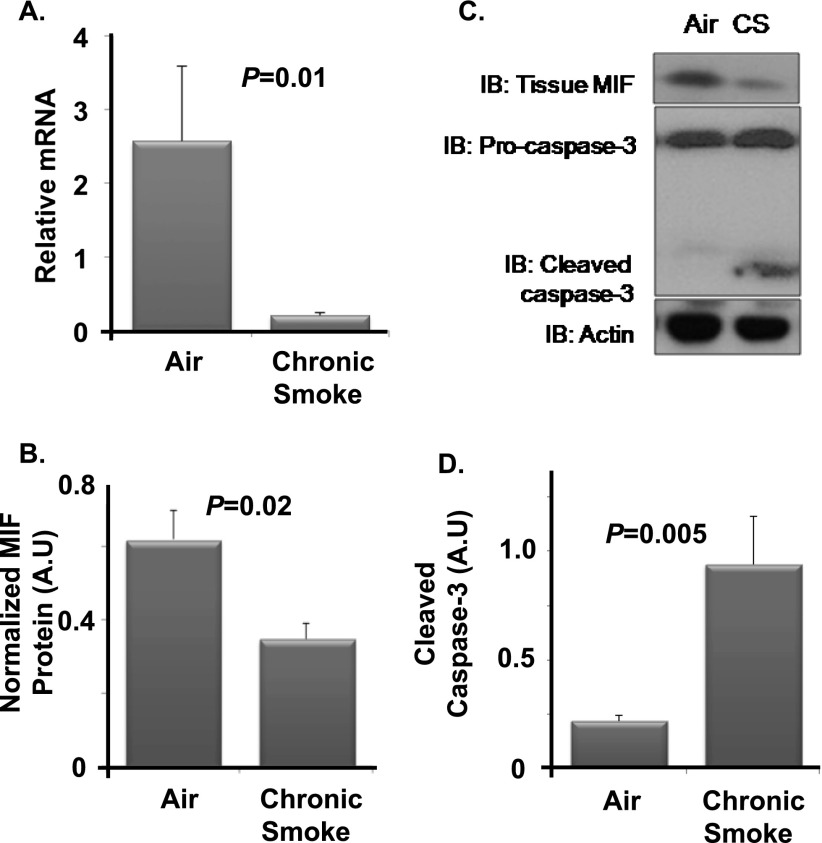
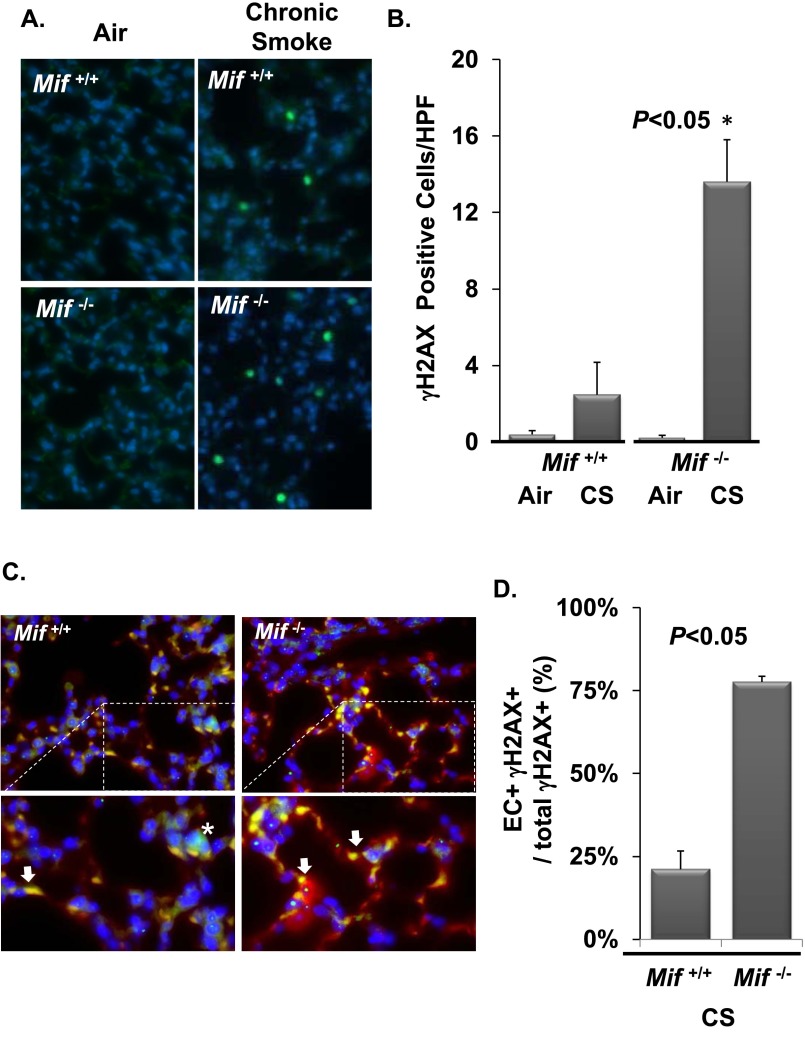
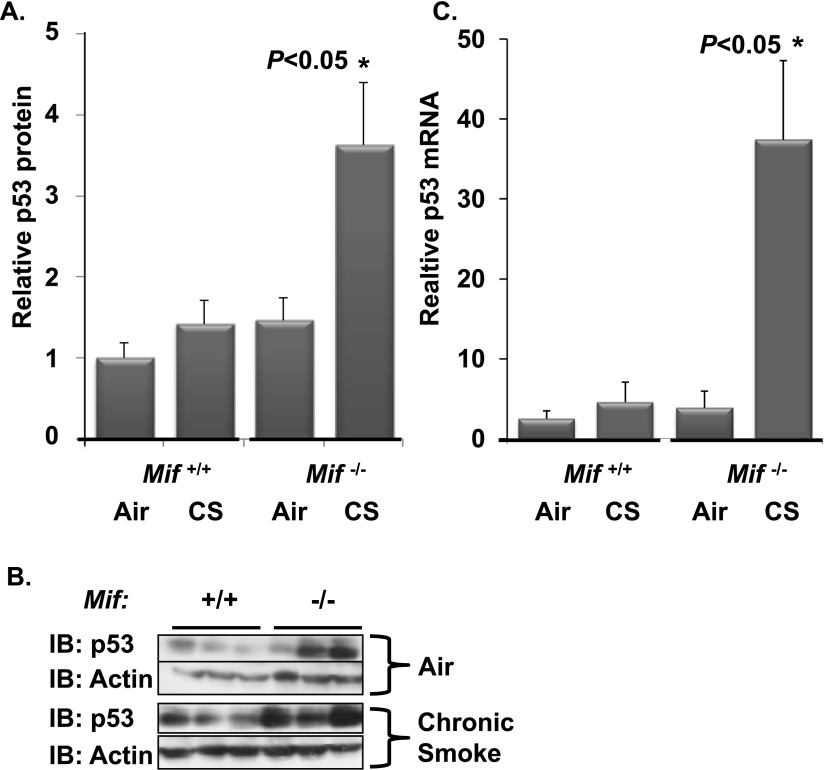
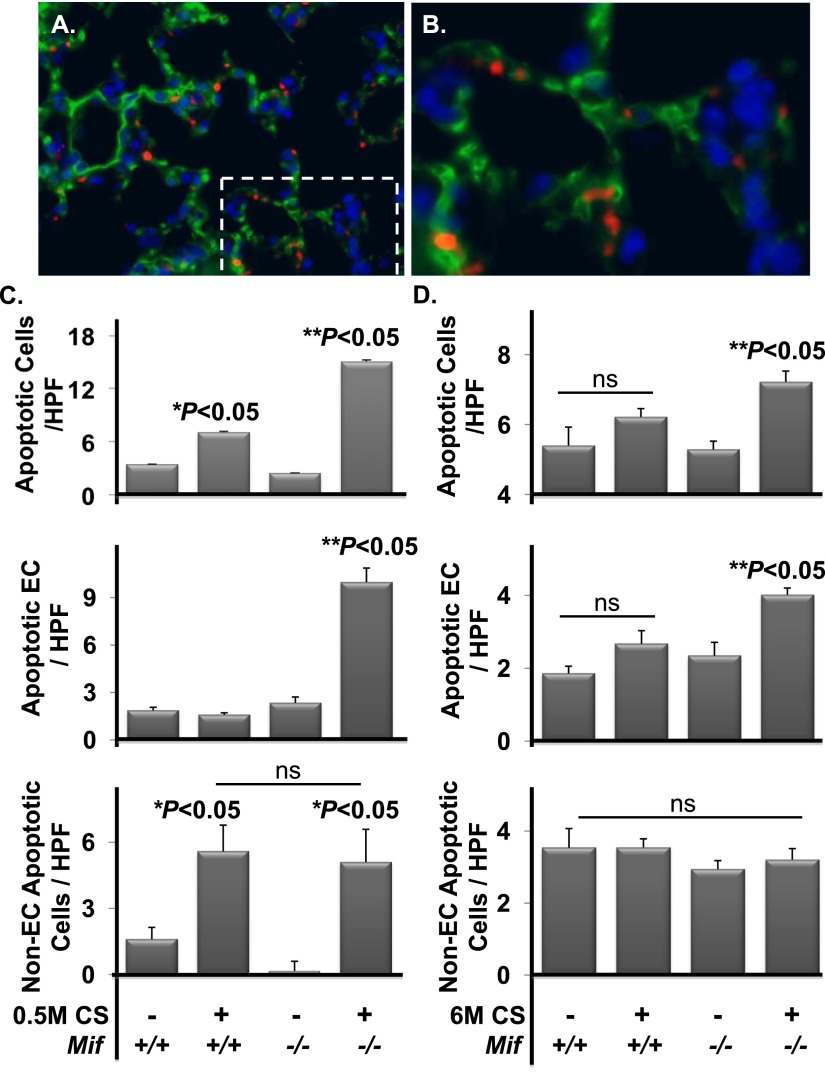
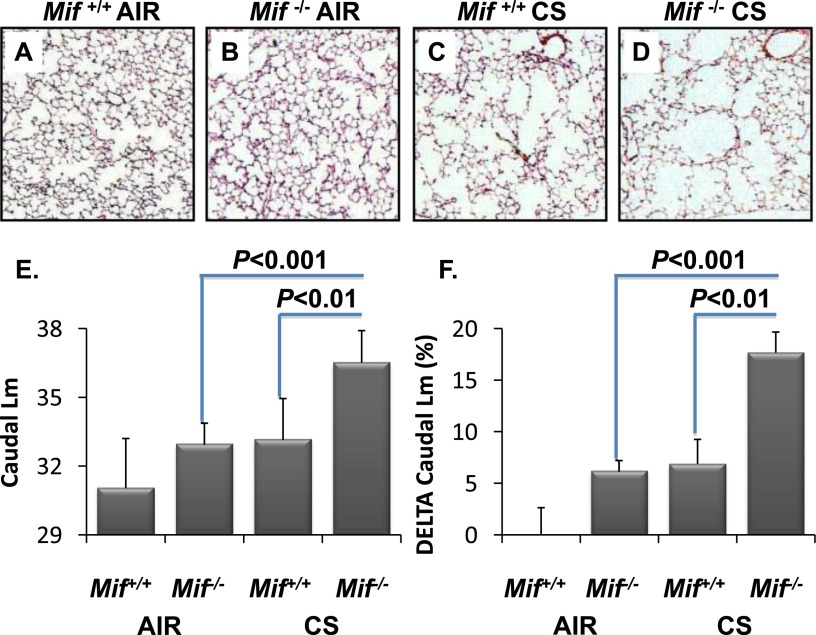
Cigarette smoke (CS) is the most common cause of chronic obstructive pulmonary diseases (COPD), including emphysema. CS exposure impacts all cell types within the airways and lung parenchyma, causing alveolar tissue destruction through four mechanisms: (1) oxidative stress; (2) inflammation; (3) protease-induced degradation of the extracellular matrix; and (4) enhanced alveolar epithelial and endothelial cell (EC) apoptosis. Studies in human pulmonary ECs demonstrate that macrophage migration inhibitory factor (MIF) antagonizes CS-induced apoptosis. Here, we used human microvascular ECs, an animal model of emphysema (mice challenged with chronic CS), and patient serum samples to address both the capacity of CS to alter MIF expression and the effects of MIF on disease severity. We demonstrate significantly reduced serum MIF levels in patients with COPD. In the murine model, chronic CS exposure resulted in decreased MIF mRNA and protein expression in the intact lung. MIF deficiency (Mif(-/-)) potentiated the toxicity of CS exposure in vivo via increased apoptosis of ECs, resulting in enhanced CS-induced tissue remodeling. This was linked to MIF's capacity to protect against double-stranded DNA damage and suppress p53 expression. Taken together, MIF appears to antagonize CS-induced toxicity in the lung and resultant emphysematous tissue remodeling by suppressing EC DNA damage and controlling p53-mediated apoptosis, highlighting a critical role of MIF in EC homeostasis within the lung.
Keywords: apoptosis; cigarette; emphysema; endothelial; macrophage migration inhibitory factor.
Figures
References
-
- Kanazawa H, Yoshikawa J. Elevated oxidative stress and reciprocal reduction of vascular endothelial growth factor levels with severity of COPD. Chest. 2005;128:3191–3197. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
