

Polycystin-1: a master regulator of intersecting cystic pathways
- PMID: 24491980
- PMCID: PMC4008641
- DOI: 10.1016/j.molmed.2014.01.004
Polycystin-1: a master regulator of intersecting cystic pathways
Abstract
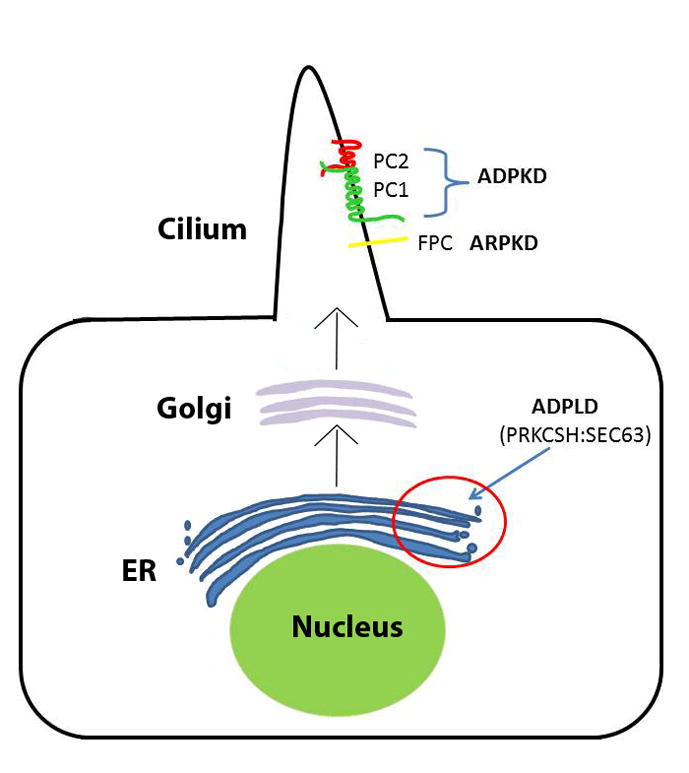
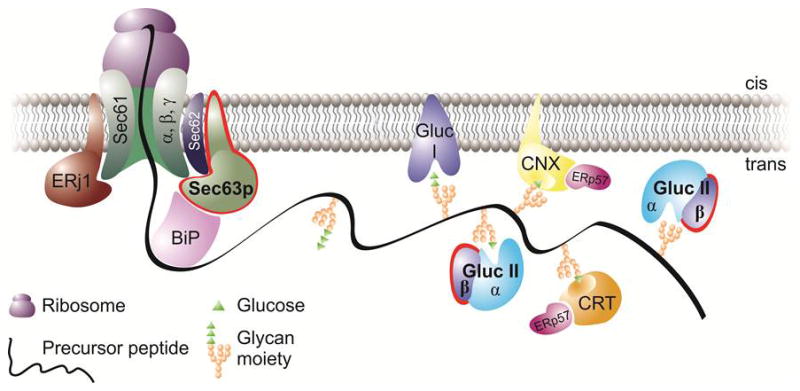
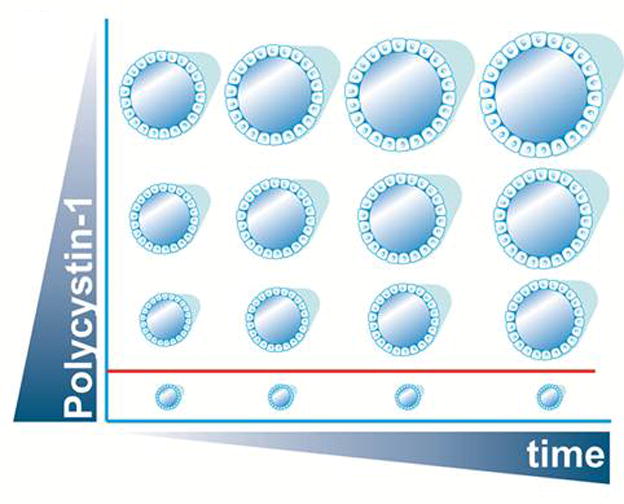
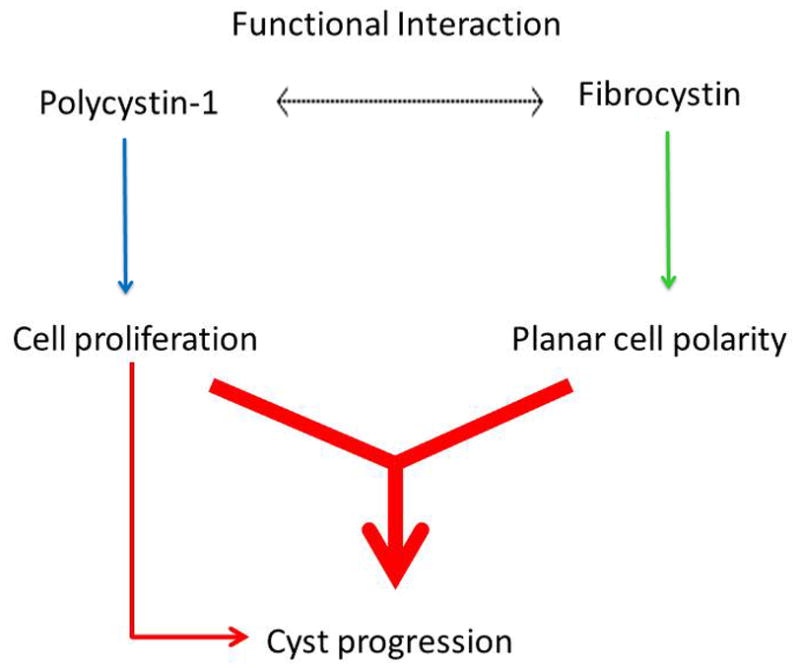
Autosomal dominant polycystic kidney disease (ADPKD) is the most common potentially lethal monogenic disorder, with more than 12 million cases worldwide. The two causative genes for ADPKD, PKD1 and PKD2, encode protein products polycystin-1 (PC1) and polycystin-2 (PC2 or TRPP2), respectively. Recent data have shed light on the role of PC1 in regulating the severity of the cystic phenotypes in ADPKD, autosomal recessive polycystic kidney disease (ARPKD), and isolated autosomal dominant polycystic liver disease (ADPLD). These studies showed that the rate for cyst growth was a regulated trait, a process that can be either sped up or slowed down by alterations in functional PC1. These findings redefine the previous understanding that cyst formation occurs as an 'on-off' process. Here, we review these and other related studies with an emphasis on their translational implications for polycystic diseases.
Keywords: chaperone therapy; cyst progression; polycystic kidney disease; polycystic liver disease; polycystin-1 dosage; protein biogenesis.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures
References
-
- Guay-Woodford LM. Renal cystic diseases: diverse phenotypes converge on the cilium/centrosome complex. Pediatr Nephrol. 2006;21:1369–1376. - PubMed
-
- Hughes J, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10:151–160. - PubMed
-
- Mochizuki T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. - PubMed
-
- Nauli SM, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. - PubMed
-
- Qian F, et al. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–987. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
