

A Bayesian sampler for optimization of protein domain hierarchies
- PMID: 24494927
- PMCID: PMC3948484
- DOI: 10.1089/cmb.2013.0099
A Bayesian sampler for optimization of protein domain hierarchies
Abstract
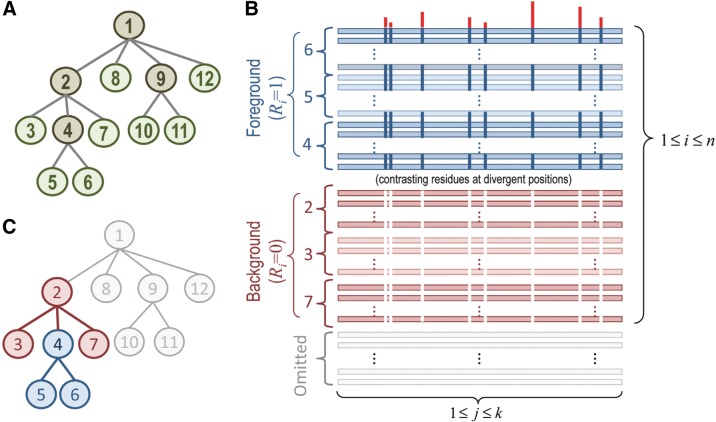
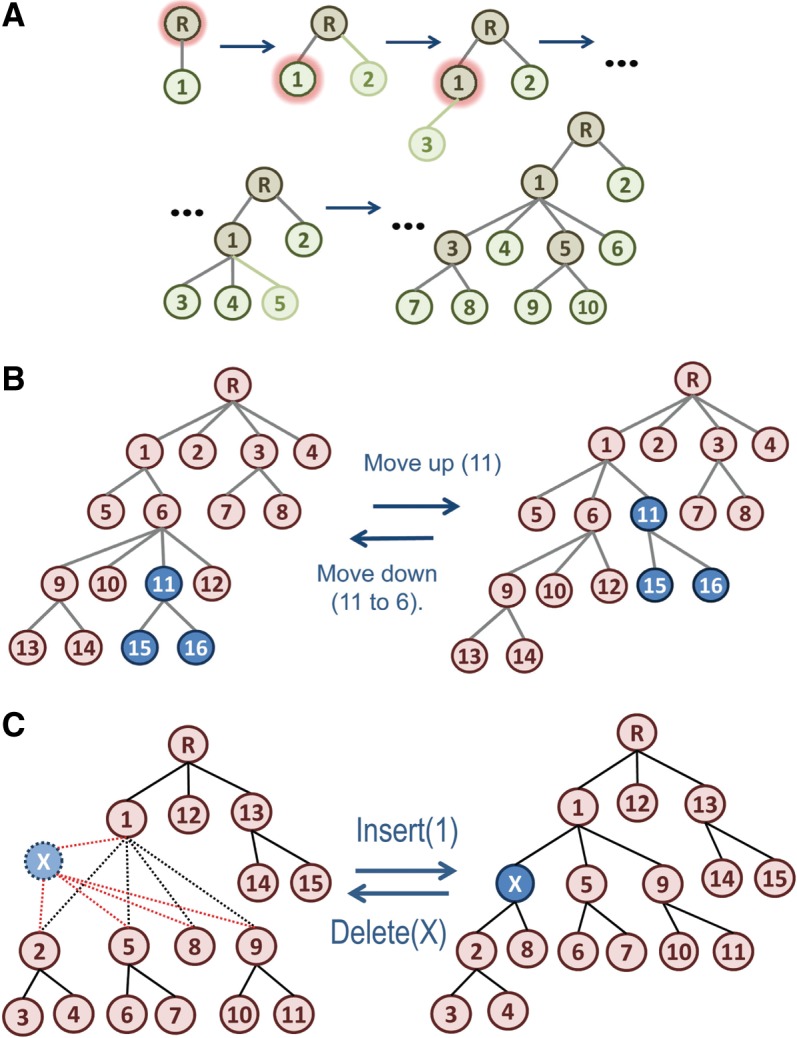
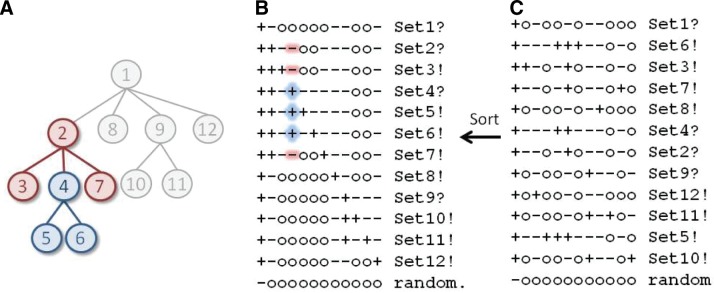
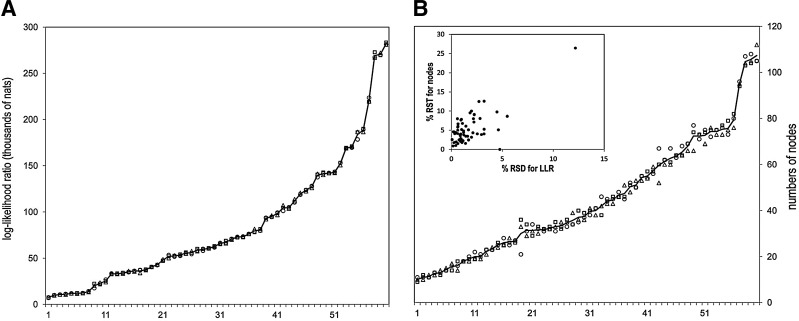
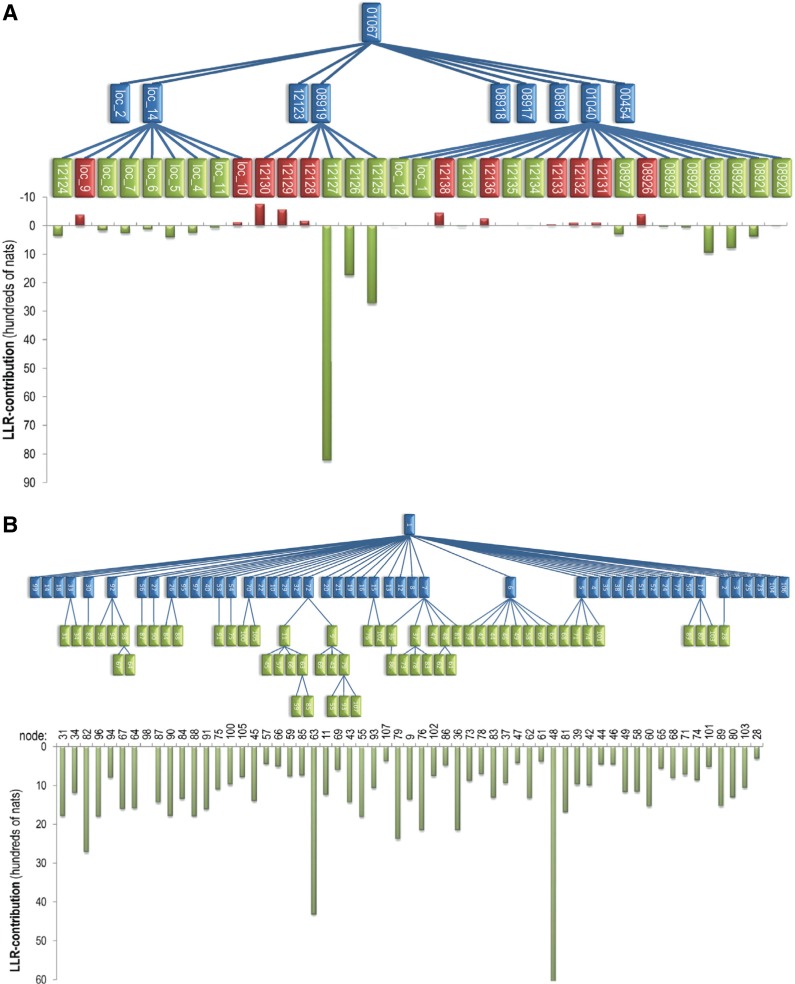
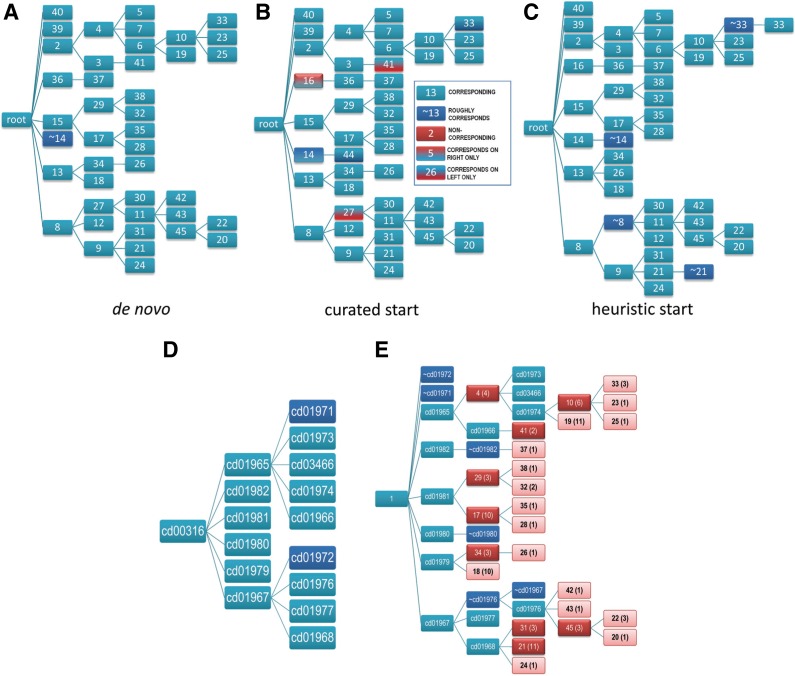
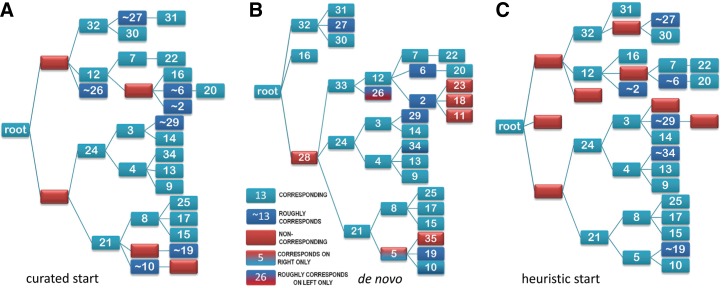
The process of identifying and modeling functionally divergent subgroups for a specific protein domain class and arranging these subgroups hierarchically has, thus far, largely been done via manual curation. How to accomplish this automatically and optimally is an unsolved statistical and algorithmic problem that is addressed here via Markov chain Monte Carlo sampling. Taking as input a (typically very large) multiple-sequence alignment, the sampler creates and optimizes a hierarchy by adding and deleting leaf nodes, by moving nodes and subtrees up and down the hierarchy, by inserting or deleting internal nodes, and by redefining the sequences and conserved patterns associated with each node. All such operations are based on a probability distribution that models the conserved and divergent patterns defining each subgroup. When we view these patterns as sequence determinants of protein function, each node or subtree in such a hierarchy corresponds to a subgroup of sequences with similar biological properties. The sampler can be applied either de novo or to an existing hierarchy. When applied to 60 protein domains from multiple starting points in this way, it converged on similar solutions with nearly identical log-likelihood ratio scores, suggesting that it typically finds the optimal peak in the posterior probability distribution. Similarities and differences between independently generated, nearly optimal hierarchies for a given domain help distinguish robust from statistically uncertain features. Thus, a future application of the sampler is to provide confidence measures for various features of a domain hierarchy.
Figures
Similar articles
-
Protein domain hierarchy Gibbs sampling strategies.Stat Appl Genet Mol Biol. 2014 Aug;13(4):497-517. doi: 10.1515/sagmb-2014-0008. Stat Appl Genet Mol Biol. 2014. PMID: 24988248
-
Automated hierarchical classification of protein domain subfamilies based on functionally-divergent residue signatures.BMC Bioinformatics. 2012 Jun 22;13:144. doi: 10.1186/1471-2105-13-144. BMC Bioinformatics. 2012. PMID: 22726767 Free PMC article.
-
Evaluating, comparing, and interpreting protein domain hierarchies.J Comput Biol. 2014 Apr;21(4):287-302. doi: 10.1089/cmb.2013.0098. Epub 2014 Feb 21. J Comput Biol. 2014. PMID: 24559108 Free PMC article.
-
Bayesian coestimation of phylogeny and sequence alignment.BMC Bioinformatics. 2005 Apr 1;6:83. doi: 10.1186/1471-2105-6-83. BMC Bioinformatics. 2005. PMID: 15804354 Free PMC article.
-
Bayesian models and Markov chain Monte Carlo methods for protein motifs with the secondary characteristics.J Comput Biol. 2005 Sep;12(7):952-70. doi: 10.1089/cmb.2005.12.952. J Comput Biol. 2005. PMID: 16201915 Review.
Cited by
-
Tracing the origin and evolution of pseudokinases across the tree of life.Sci Signal. 2019 Apr 23;12(578):eaav3810. doi: 10.1126/scisignal.aav3810. Sci Signal. 2019. PMID: 31015289 Free PMC article.
-
Deep Analysis of Residue Constraints (DARC): identifying determinants of protein functional specificity.Sci Rep. 2020 Feb 3;10(1):1691. doi: 10.1038/s41598-019-55118-6. Sci Rep. 2020. PMID: 32015389 Free PMC article.
-
Inferring joint sequence-structural determinants of protein functional specificity.Elife. 2018 Jan 16;7:e29880. doi: 10.7554/eLife.29880. Elife. 2018. PMID: 29336305 Free PMC article.
-
Bayesian Top-Down Protein Sequence Alignment with Inferred Position-Specific Gap Penalties.PLoS Comput Biol. 2016 May 18;12(5):e1004936. doi: 10.1371/journal.pcbi.1004936. eCollection 2016 May. PLoS Comput Biol. 2016. PMID: 27192614 Free PMC article.
-
Evolution of Functional Diversity in the Holozoan Tyrosine Kinome.Mol Biol Evol. 2021 Dec 9;38(12):5625-5639. doi: 10.1093/molbev/msab272. Mol Biol Evol. 2021. PMID: 34515793 Free PMC article.
References
-
- Abascal F., and Valencia A.2002. Clustering of proximal sequence space for the identification of protein families. Bioinformatics 18, 908–921 - PubMed
-
- Hastings W.K.1970. Monte Carlo sampling methods using Markov Chains and their applications. Biometrika 57, 97–109
-
- Kirkpatrick S., Gelatt C.D., and Vecchi M.P.1983. Optimization by simulated annealing. Science 220, 671–680 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
