

Constructing module maps for integrated analysis of heterogeneous biological networks
- PMID: 24497192
- PMCID: PMC3985673
- DOI: 10.1093/nar/gku102
Constructing module maps for integrated analysis of heterogeneous biological networks
Abstract
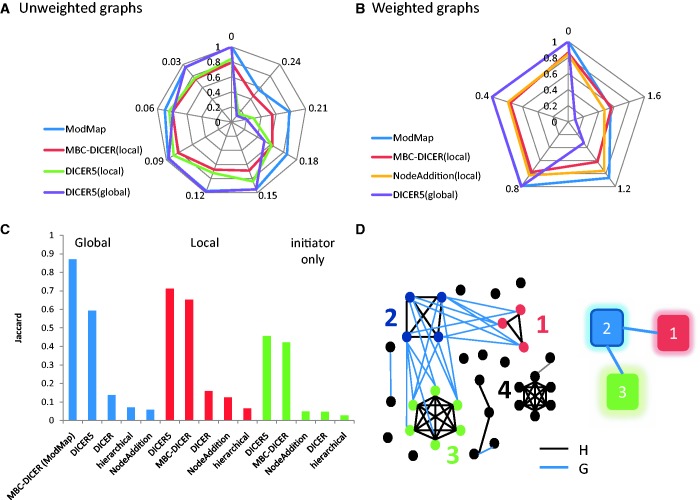
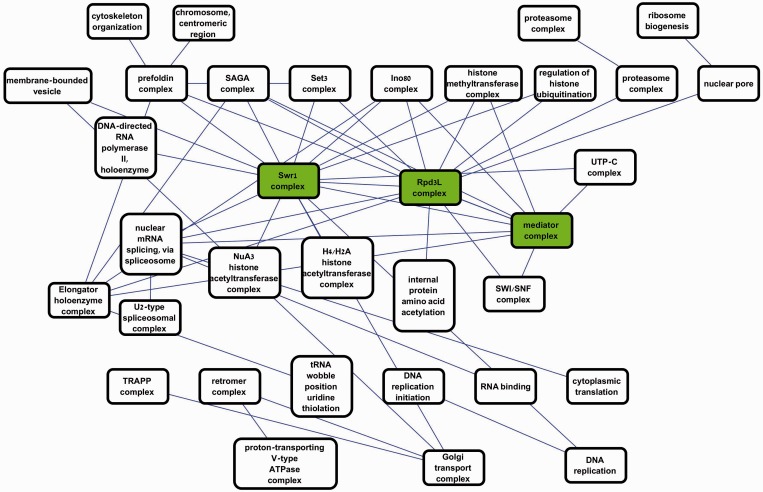
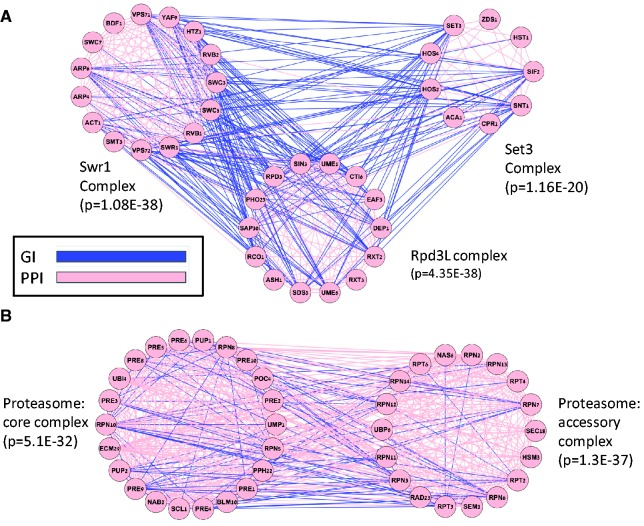
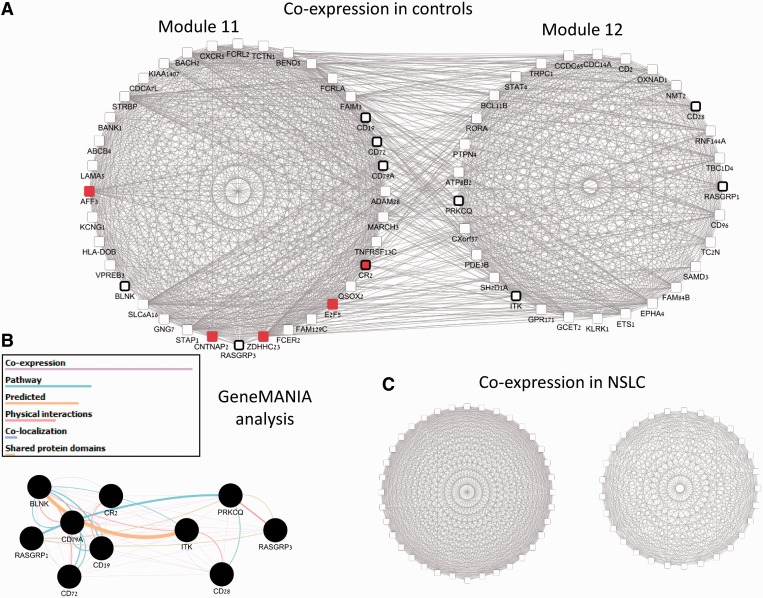
Improved methods for integrated analysis of heterogeneous large-scale omic data are direly needed. Here, we take a network-based approach to this challenge. Given two networks, representing different types of gene interactions, we construct a map of linked modules, where modules are genes strongly connected in the first network and links represent strong inter-module connections in the second. We develop novel algorithms that considerably outperform prior art on simulated and real data from three distinct domains. First, by analyzing protein-protein interactions and negative genetic interactions in yeast, we discover epistatic relations among protein complexes. Second, we analyze protein-protein interactions and DNA damage-specific positive genetic interactions in yeast and reveal functional rewiring among protein complexes, suggesting novel mechanisms of DNA damage response. Finally, using transcriptomes of non-small-cell lung cancer patients, we analyze networks of global co-expression and disease-dependent differential co-expression and identify a sharp drop in correlation between two modules of immune activation processes, with possible microRNA control. Our study demonstrates that module maps are a powerful tool for deeper analysis of heterogeneous high-throughput omic data.
Figures

References
-
- Deng MH, Zhang K, Mehta S, Chen T, Sun FZ. Prediction of protein function using protein-protein interaction data. J. Comput. Biol. 2003;10:947–960. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
